
Abstract
Background:
Prucalopride (1 or 2 mg once daily) is approved for treating adults with chronic idiopathic constipation (CIC).
Objectives:
We determined the effect of age, body mass index (BMI), and renal function on the efficacy and safety of prucalopride in adults with CIC.
Design:
Data were pooled from six 12-week, phase III–IV clinical studies in adults who received prucalopride (1 or 2 mg once daily) or placebo for CIC.
Methods:
Adults were stratified by age (<50; 50–64; ⩾65 years), BMI (underweight/healthy weight, <25 kg/m2; overweight, 25 to <30 kg/m2; obese, ⩾30 kg/m2), and renal function (normal renal function, estimated glomerular filtration rate (eGFR) ⩾90 mL/min/1.73 m2; mild renal impairment, eGFR 60 to <90 mL/min/1.73 m2; moderate renal impairment, eGFR 30 to <60 mL/min/1.73 m2). The primary efficacy endpoint was the proportion of patients with a mean of ⩾3 complete spontaneous bowel movements/week over 12 weeks. Safety data were evaluated descriptively.
Results:
Of 2484 patients stratified by age (prucalopride, n = 1237; placebo, n = 1247), 1402, 708, and 374 were aged <50, 50–64, and ⩾65 years, respectively. Of 2482 patients stratified by BMI (prucalopride, n = 1237; placebo, n = 1245), 1425, 713, and 344 were underweight/healthy weight, overweight, and obese, respectively. Of 2474 patients stratified by renal function (prucalopride, n = 1233; placebo, n = 1241), 1444, 869, and 161 had normal renal function, mild renal impairment, and moderate renal impairment, respectively. More prucalopride-treated than placebo-treated patients achieved the primary efficacy endpoint. The difference was significant for all subgroups, except for the obese and moderate renal impairment subgroups. More prucalopride-treated than placebo-treated patients reported treatment-related adverse events in most subgroups.
Conclusion:
Prucalopride demonstrated efficacy in adults with CIC, irrespective of age, BMI, and renal function. No unexpected safety concerns were identified.
Trial registration:
ClinicalTrials.gov identifiers (https://clinicaltrials.gov/): NCT01147926, NCT01424228, NCT01116206, NCT00483886, NCT00485940, NCT00488137.
Introduction
Chronic idiopathic constipation (CIC) is a functional bowel disorder that is prevalent globally and affects 14% of individuals aged 15 years or older. 1 The condition is broadly characterized by a reduced stool frequency of less than three stools per week, difficult defecation, or a feeling of having an incomplete bowel movement. 2
Although the condition can affect individuals of all demographics, the prevalence of CIC is higher among women than men and increases with age in those over 50 years old, with the highest prevalence in those over 70 years old. 3 Some studies have also suggested that there is a higher prevalence of constipation in obese individuals than in individuals with a healthy body weight.4–6 Obesity is not thought to be a direct cause of constipation, although other factors potentially related to obesity, such as activity levels and/or diet, could contribute to constipation.7,8 However, evidence does suggest that obesity may contribute to varied drug metabolism, leading to potential underdosing or overdosing, which may impact clinical efficacy. 9 Therefore, it is important to understand how treatments for CIC are affected by age and body weight, particularly because the prevalence of obesity is increasing worldwide. 10
Renal excretion is the main route of prucalopride elimination; on average, 84.2% and 13.3% of the administered dose is recovered in the urine and feces, respectively, of healthy individuals.11,12 Given that prucalopride is predominantly excreted by the kidneys, a reduced oral dosing regimen (1 mg once daily) is indicated for adult patients with severe renal impairment (creatinine clearance <30 mL/min/1.73 m 2 ) in the USA and Europe.11,13 The effect of renal function on prucalopride pharmacokinetics has been evaluated in a phase I study in participants without CIC who had either normal renal function or moderate/severe renal impairment. Significant reductions in the renal clearance of prucalopride were observed in participants with moderate or severe renal impairment compared to those with normal renal function. 14 Constipation is one of the gastrointestinal comorbidities most commonly associated with chronic kidney disease,15,16 with an estimated prevalence of 37 million people in the USA alone in 2021. 17 Therefore, it is important to understand the impact of renal impairment on the efficacy and safety of prucalopride in patients with CIC.
Current treatment guidelines from the American Gastroenterological Association and the American College of Gastroenterology (2023) for CIC in adults recommend changes to lifestyle and diet, such as increasing fiber and fluid intake. 18 If a patient does not respond to lifestyle and dietary modifications, then osmotic and stimulant laxatives, secretagogues, and prokinetic agents can be used to improve bowel function.18–20 The US Food and Drug Administration approved prucalopride, a selective serotonin type 4 receptor agonist with prokinetic activity, for the treatment of CIC in adults (2 mg once daily; or 1 mg once daily in patients with severe renal impairment). Prucalopride became available to US patients on April 2, 2019.11,21 In Europe, prucalopride has been approved since October 15, 2009 for managing chronic constipation symptoms in adults for whom laxatives have failed to provide adequate relief.13,22 Prucalopride (1 or 2 mg once daily) has been demonstrated to improve the number of complete spontaneous bowel movements (CSBMs) in patients with CIC over a 12-week treatment period as part of an integrated analysis of six phase III and IV clinical trials. 23 In this same analysis, the efficacy and safety data for prucalopride have been summarized and stratified by sex. 23 The efficacy and safety endpoints were not significantly different between male and female patients. 23 A pharmacokinetic analysis of prucalopride showed that there are no clinically significant differences in its pharmacokinetic profile based on age (17–95 years) or body mass index (BMI; 14–57 kg/m2) after accounting for the effect of renal function. 11 However, efficacy and safety endpoints were not analyzed. This information would help to improve clinical understanding and enable healthcare professionals to tailor therapies based on individual patients’ characteristics.
This post hoc analysis therefore aimed to investigate the effect of age, BMI, and renal function on the efficacy and safety of prucalopride in patients with CIC.
Methods
Study design and patients
In this post hoc analysis, data were collected from six key phase III and IV multicenter, double-blind, randomized, placebo-controlled clinical studies of prucalopride (1 or 2 mg once daily for 12 weeks) in patients with CIC (ClinicalTrials.gov identifiers: NCT01147926 (SPD555-302), 24 NCT01424228 (SPD555-401), 25 NCT01116206 (PRU-CRC-3001), 26 NCT00483886 (PRU-USA-11), 27 NCT00485940 (PRU-USA-13), 28 and NCT00488137 (PRU-INT-6) 29 ). The US Food and Drug Administration-approved dosage of prucalopride for CIC is 2 mg once daily in adults without severe renal impairment or 1 mg once daily in adults with severe renal impairment. 11 Only patients who received the approved dosage of prucalopride for CIC were included in this analysis. In two of the studies, patients aged 65 years or older received a dose of 1 mg, which is the approved dose for this age group in Europe.13,23 Independent institutional review boards or independent ethics committees approved the studies, which were conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines, and relevant regulatory requirements. Patients provided written informed consent before participating in the studies.
Patients were included if they had one or more of the following for at least 6 months: two or fewer CSBMs per week; hard or very hard stools; a feeling of incomplete evacuation; or straining during defecation in at least 25% of bowel movements.24–29 Other key inclusion criteria included: male or female (nonpregnant, non-breastfeeding) patients who were aged 18 years or older (an upper age limit of 65 years was specified in only one of the studies) 26 ; patients who were willing and able to fill out a patient diary and questionnaires without help; and patients who were available for follow-up (ClinicalTrials.gov identifiers: NCT01147926, NCT01424228, NCT01116206, NCT00483886, NCT00485940, NCT00488137).
Exclusion criteria included drug-induced constipation; constipation secondary to causes such as endocrine, metabolic, and neurological disorders, or surgery; a history of clinically significant cancer or cardiac, vascular, hepatic, pulmonary, endocrine, metabolic, neurological, or psychiatric disorders; known or suspected disorders of the large bowel (obstruction, carcinoma, or inflammatory bowel disease); previous use of prucalopride or any other investigational drug in the 30 days before the screening visit; and clinically significant abnormalities of hematology, urinalysis, or blood chemistry as determined by the investigator (ClinicalTrials.gov identifiers: NCT01147926, NCT01424228, NCT01116206, NCT00483886, NCT00485940, NCT00488137). Patients with severe renal impairment (creatinine clearance <30 mL/min/1.73 m2) were excluded.24–29 Additional inclusion and exclusion criteria are reported in previously published literature.24–29
In this post hoc analysis, adult patients were stratified by:
age into three subgroups (<50; 50–64; or ⩾65 years).
BMI into three subgroups (underweight/healthy weight, <25 kg/m2; overweight, 25 to <30 kg/m2; or obese, ⩾30 kg/m2; BMI categories are based on the Centers for Disease Control and Prevention classification). 30
estimated glomerular filtration rate into three renal function subgroups (normal renal function, ⩾90 mL/min/1.73 m2; mild renal impairment, 60 to <90 mL/min/1.73 m2; or moderate renal impairment, 30 to <60 mL/min/1.73 m2).
Efficacy endpoints
The prespecified primary efficacy endpoint for all six studies was defined as the proportion of patients with a mean frequency of at least three CSBMs per week over 12 weeks. An alternative and more stringent primary efficacy endpoint was defined as the proportion of patients with a mean frequency of at least three CSBMs per week over 12 weeks and an increase of at least one CSBM per week from baseline in at least 9 out of the 12 weeks, including 3 of the last 4 weeks, and was evaluated post hoc. This efficacy endpoint was prespecified in only one of the six clinical studies (SPD555-302) and was included as a secondary efficacy endpoint.
The prespecified secondary efficacy endpoints for all six studies were analyzed from baseline to weeks 1–12 of treatment and included the following: change in CSBM frequency; time to first CSBM; change in stool characteristics (proportion of stools with a normal consistency or a hard to very hard consistency); change in bowel movements (proportion of bowel movements with no straining or with severe/very severe straining); change in rescue medication use; and changes in Patient Assessment of Constipation Symptoms questionnaire (PAC-SYM) and Patient Assessment of Constipation Quality of Life questionnaire (PAC-QOL) total scores. Changes in the global severity of constipation and efficacy of treatment scores were also measured at week 12 of treatment together with the proportion of responders and nonresponders at baseline and week 12 of treatment where appropriate.
Safety endpoints
Safety endpoints were also analyzed over the 12-week treatment period. Any adverse events (AEs) that occurred after the first dose of prucalopride or placebo were considered treatment-emergent AEs (TEAEs) and were graded according to their severity (mild, moderate, or severe). Any TEAEs considered at least possibly related to the study drug were defined as treatment-related TEAEs. Cardiovascular (CV) events of interest (angina pectoris, angina unstable, cerebrovascular accident, ischemic stroke, myocardial infarction, and myocardial ischemia) were also included in this post hoc analysis.
Statistical analyses
Efficacy analyses were performed using the full analysis set, which included all patients who received at least one dose of the study drug and who had at least one efficacy assessment after receiving the initial dose. The primary efficacy endpoint was compared between prucalopride-treated and placebo-treated patients, stratified by age, BMI, and renal function, using the χ 2 test. The change in CSBM frequency was assessed using a Cochran–Mantel–Haenszel test, and the time to first CSBM was evaluated using a proportional hazards regression model (both analyses were controlled for clinical study number, patient sex, country, and number of complete bowel movements per week at baseline (0 or >0)). Other secondary efficacy endpoints were assessed using an analysis of covariance model, including the treatment group, study, country, patient sex, and number of complete bowel movements per week at baseline (0 or >0) as factors and the baseline endpoint measure as a covariate. Safety analyses were performed using the safety analysis set, which included all patients who received at least one dose of the study drug. Safety data were evaluated using descriptive statistics.
Results
Patient demographics and clinical characteristics at baseline
In total, 2484 patients (prucalopride, n = 1237; placebo, n = 1247), 2482 patients (prucalopride, n = 1237; placebo, n = 1245), and 2474 patients (prucalopride, n = 1233; placebo, n = 1241) with available data were stratified by age, BMI, and renal function, respectively (Figure 1). Baseline patient demographics and clinical characteristics by treatment group and stratified by age, BMI, and renal function are presented in Tables 1–3, respectively. In the overall population, 1402 patients (56.4%) were aged younger than 50 years, 708 (28.5%) were aged 50–64 years, and 374 (15.1%) were aged 65 years or older. Most of the patients aged younger than 50 or 50–64 years were female, and the majority of patients aged 65 years or older were male. Across all age subgroups, most patients were White. The mean (standard deviation (SD)) duration of constipation was 13.7 (11.0), 20.4 (16.0), and 19.6 (20.4) years for patients aged younger than 50, 50–64, and 65 years or older, respectively. Overall, 27.7%, 35.2%, and 29.1% of patients aged younger than 50, 50–64, and 65 years or older, respectively, had no spontaneous bowel movements (SBMs) at baseline.

CONSORT flow diagram.
Patient demographics and clinical characteristics at baseline, stratified by age.

Less than 50 years: prucalopride, n = 706; placebo, n = 695; 50–64 years: prucalopride, n = 335; placebo, n = 372; ⩾65 years: prucalopride, n = 196; placebo, n = 178.
SBMs per week were measured during the 6-month period before study initiation.
Less than 50 years: prucalopride, n = 690; placebo, n = 677; 50–64 years: prucalopride, n = 323; placebo, n = 360; ⩾65 years: prucalopride, n = 188; placebo, n = 174.
BMI, body mass index; SBM, spontaneous bowel movement; SD, standard deviation.
Patient demographics and clinical characteristics at baseline, stratified by BMI.

BMI was classified according to the Centers for Disease Control and Prevention classification. 17 Underweight/healthy weight, BMI < 25 kg/m2; overweight, BMI 25 to <30 kg/m2; obese, BMI ⩾30 kg/m2.
SBMs per week were measured during the 6-month period before study initiation.
Underweight/healthy weight: prucalopride, n = 663; placebo, n = 725; overweight: prucalopride, n = 378; placebo, n = 317; obese: prucalopride, n = 160; placebo, n = 167.
BMI, body mass index; SBM, spontaneous bowel movement; SD, standard deviation.
Patient demographics and clinical characteristics at baseline, stratified by renal function.

Normal renal function, eGFR ⩾90 mL/min/1.73 m2; mild renal impairment, eGFR 60 to <90 mL/min/1.73 m2; moderate renal impairment, eGFR 30 to <60 mL/min/1.73 m2.
SBMs per week were measured during the 6-month period before clinical study initiation.
Normal renal function: prucalopride, n = 696; placebo, n = 697; mild renal impairment: prucalopride, n = 423; placebo, n = 427; moderate renal impairment: prucalopride, n = 78; placebo, n = 81.
eGFR, estimated glomerular filtration rate; SBM, spontaneous bowel movement; SD, standard deviation.
In the overall population, 90 patients (3.6%) were underweight, 1335 (53.8%) were a healthy weight, 713 (28.7%) were overweight, and 344 (13.9%) were obese. Owing to the small number of underweight patients, this subgroup was combined with patients with a healthy weight (forming the “underweight/healthy weight” subgroup). The mean (SD) age was 43.8 (15.1), 52.6 (15.2), and 52.0 (13.9) years for underweight/healthy weight, overweight, and obese patients, respectively. Across all BMI subgroups, most patients were female and White. The mean (SD) duration of constipation was 16.2 (14.1), 16.8 (15.5), and 17.0 (14.9) years for underweight/healthy weight, overweight, and obese patients, respectively. Overall, 30.4%, 29.2%, and 30.2% of underweight/healthy weight, overweight, and obese patients, respectively, had no SBMs at baseline.
In the overall population, 1444 patients (58.4%) had normal renal function, 869 (35.1%) had mild renal impairment, and 161 (6.5%) had moderate renal impairment. The mean (SD) age was 42.5 (13.0), 52.1 (15.6), and 66.7 (13.9) years for patients in the normal renal function, mild renal impairment, and moderate renal impairment subgroups, respectively. Across all renal function subgroups, most patients were female and White. The mean (SD) duration of constipation was 15.4 (13.5), 17.1 (15.1), and 21.9 (19.0) years for patients in the normal renal function, mild renal impairment, and moderate renal impairment subgroups, respectively. Overall, 27.6%, 32.8%, and 36.0% of patients in the normal renal function, mild renal impairment, and moderate renal impairment subgroups had no SBMs at baseline.
Of the 1237 prucalopride-treated patients in the age and BMI subgroups, only 21 patients received prucalopride 1 mg once daily and 1216 received prucalopride 2 mg once daily. Of the 1233 prucalopride-treated patients in the renal function subgroups, 21 patients received prucalopride 1 mg once daily and 1212 patients received prucalopride 2 mg once daily. A stratification analysis by prucalopride dose was not performed owing to the small number of patients receiving prucalopride 1 mg. Baseline demographics and clinical characteristics were generally similar for patients in the prucalopride-treated and placebo-treated groups stratified either by age, BMI, or renal function (Tables 1–3, respectively).
Efficacy endpoints
Prespecified primary efficacy endpoint
Within each age subgroup, a significantly greater proportion of prucalopride-treated than placebo-treated patients achieved a mean frequency of at least three CSBMs per week over 12 weeks of treatment (<50 years: 26.3% vs 11.4%, p < 0.001; 50–64 years: 31.0% vs 15.0%, p < 0.001; ⩾65 years: 28.1% vs 16.9%, p = 0.010; Figure 2(a)).

Prespecified and post hoc primary efficacy endpoints. Proportions of prucalopride-treated and placebo-treated patients with a mean frequency of at least three CSBMs per week over 12 weeks of treatment (prespecified), and a mean frequency of at least three CSBMs per week over 12 weeks and an increase of at least one CSBM per week from baseline in at least 9 out of the 12 weeks, including 3 of the last 4 weeks (post hoc), stratified by age (aa and dd, respectively), BMI (bb and ee, respectively), and renal function (cc and ff, respectively). BMI was classified according to the Centers for Disease Control and Prevention classification. 17 Underweight/healthy weight, BMI <25 kg/m2; overweight, BMI 25 to <30 kg/m2; obese, BMI ⩾30 kg/m2. Normal renal function, eGFR ⩾90 mL/min/1.73 m2; mild renal impairment, eGFR 60 to <90 mL/min/1.73 m2. p values are based on the χ2 test.
Significantly more prucalopride-treated than placebo-treated patients achieved a mean of at least three CSBMs per week over 12 weeks of treatment in the underweight/healthy weight subgroup (24.8% vs 10.9%, p < 0.001) and in the overweight subgroup (34.6% vs 15.4%, p < 0.001; Figure 2(b)). The proportion of obese patients who met the primary efficacy endpoint was not statistically different between the prucalopride and placebo groups, but numerically more patients in the prucalopride group than in the placebo group met the primary efficacy endpoint (25.1% vs 19.1%, p = 0.175; Figure 2(b)).
Within each renal function subgroup, a greater proportion of prucalopride-treated than placebo-treated patients achieved a mean frequency of at least three CSBMs per week over 12 weeks. However, the difference was statistically significant only for patients with normal renal function and those with mildly impaired renal function (normal renal function, 29.8% vs 13.7%, p < 0.001; mild renal impairment, 26.2% vs 12.8%, p < 0.001; moderate renal impairment, 17.7% vs 12.2%, p = 0.325; Figure 2(c)).
Post hoc primary efficacy endpoint
Within each age subgroup, a significantly greater proportion of prucalopride-treated than placebo-treated patients achieved a mean frequency of at least three CSBMs per week over 12 weeks and an increase of at least one CSBM per week from baseline in at least 9 out of the 12 weeks, including 3 of the last 4 weeks of treatment (<50 years, 18.1% vs 7.6%, p < 0.001; 50–64 years, 25.6% vs 10.4%, p < 0.001; ⩾65 years, 24.3% vs 11.8%, p = 0.002; Figure 2(d)).
Significantly more prucalopride-treated than placebo-treated patients achieved a mean frequency of at least three CSBMs per week over 12 weeks and an increase of at least one CSBM per week from baseline in at least 9 out of the 12 weeks, including 3 of the last 4 weeks of treatment, in the underweight/healthy weight subgroup (18.1% vs 7.6%, p < 0.001) and the overweight subgroup (27.5% vs 10.7%, p < 0.001; Figure 2(e)). The proportions of obese patients who met the alternative primary efficacy endpoint were not statistically different between the prucalopride and placebo groups, but numerically more obese patients in the prucalopride group than in the placebo group met the alternative primary efficacy endpoint (Figure 2(e)).
Within each renal function subgroup, a greater proportion of prucalopride-treated than placebo-treated patients achieved a mean frequency of at least three CSBMs per week over 12 weeks and an increase of at least one CSBM per week from baseline in at least 9 out of the 12 weeks, including 3 of the last 4 weeks of treatment. However, the difference was statistically significant only for patients with normal renal function and those with mildly impaired renal function (Figure 2(f)).
Prespecified secondary efficacy endpoints
Significantly more prucalopride-treated than placebo-treated patients exhibited an increase in CSBM frequency of at least one per week from baseline to week 12 of treatment across all age subgroups examined (<50 years, p < 0.001; 50–64 years, p < 0.001; ⩾65 years, p = 0.043; Figure 3(a)). Additionally, prucalopride-treated patients of all ages had a shorter time to first CSBM than placebo-treated patients (<50 years, p < 0.001; 50–64 years, p < 0.001; ⩾65 years, p = 0.003; Figure 3(b)). Other secondary efficacy endpoints in patients stratified by age are summarized in Table 4; for almost all of these efficacy endpoints, patients experienced greater improvements from baseline to weeks 1–12 with prucalopride than with placebo. The global severity of constipation and efficacy of treatment scores improved in prucalopride-treated compared with placebo-treated patients of all ages at week 12 of treatment (Supplemental Table S1).
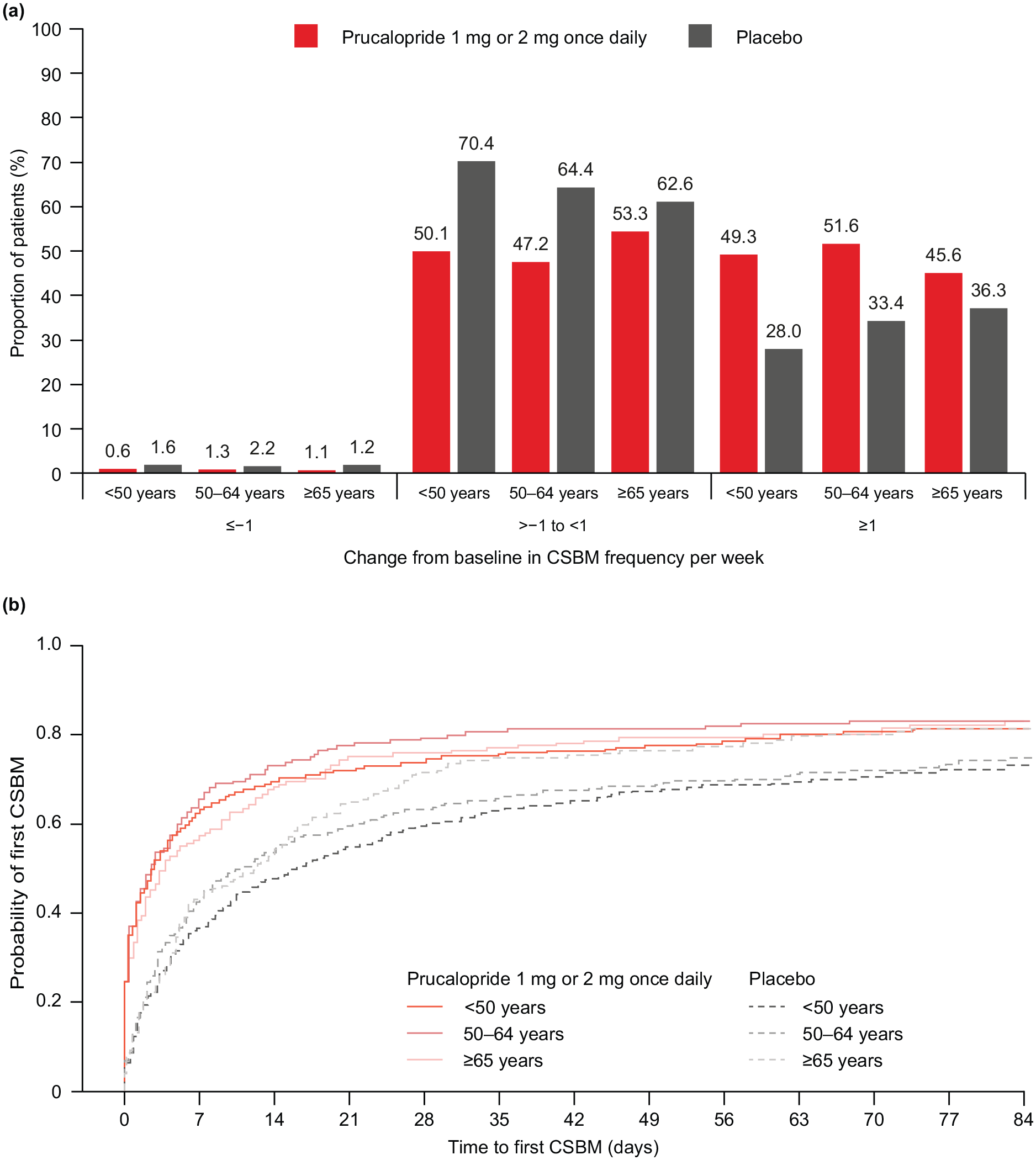
Change from baseline to week 12 in CSBM frequency per week in prucalopride-treated and placebo-treated patients (a),a and the time to first CSBM after the first dose of prucalopride or placebo (b),b stratified by age.
Summary of the secondary efficacy endpoints in prucalopride-treated and placebo-treated patients, stratified by age.

CI, confidence interval; LS, least-squares; n/a, not applicable; PAC-QOL, Patient Assessment of Constipation Quality of Life questionnaire; PAC-SYM, Patient Assessment of Constipation Symptoms questionnaire; SD, standard deviation; SE, standard error.
Significantly more prucalopride-treated than placebo-treated patients exhibited an increase in CSBM frequency of at least 1 per week from baseline to week 12 of treatment in the underweight/healthy weight (p < 0.001) and overweight (p < 0.001) subgroups (Figure 4(a)). The proportion of patients who experienced an increase in CSBM frequency from baseline to week 12 of treatment was not significantly different between obese prucalopride-treated and placebo-treated patients (p = 0.134; Figure 4(a)). Underweight/healthy weight, overweight, and obese prucalopride-treated patients had a shorter time to first CSBM than placebo-treated patients (p < 0.001, p < 0.001, and p = 0.004, respectively; Figure 4(b)). Other secondary efficacy endpoints in patients stratified by BMI are summarized in Table 5; for all of these efficacy endpoints, patients experienced greater improvements from baseline to weeks 1–12 with prucalopride than with placebo. The global severity of constipation and efficacy of treatment scores also improved in prucalopride-treated compared to placebo-treated patients across all BMI subgroups at week 12 of treatment (Supplemental Table S2).

Change from baseline to week 12 in CSBM frequency per week in prucalopride-treated and placebo-treated patients (a),a and the time to first CSBM after the first dose of prucalopride or placebo (b),b stratified by BMI. BMI was classified according to the Centers for Disease Control and Prevention classification. 17 Underweight/healthy weight, BMI <25 kg/m2; overweight, BMI 25 to <30 kg/m2; obese, BMI ⩾30 kg/m2.
Summary of the secondary efficacy endpoints in prucalopride-treated and placebo-treated patients, stratified by BMI.

BMI was classified according to the Centers for Disease Control and Prevention classification. 17 Underweight/healthy weight, BMI <25 kg/m2; overweight, BMI 25 to <30 kg/m2; obese, BMI ⩾30 kg/m2.
BMI, body mass index; CI, confidence interval; LS, least-squares; n/a, not applicable; PAC-QOL, Patient Assessment of Constipation Quality of Life questionnaire; PAC-SYM, Patient Assessment of Constipation Symptoms questionnaire; SD, standard deviation; SE, standard error.
Significantly more prucalopride-treated than placebo-treated patients exhibited an increase in CSBM frequency of at least 1 per week from baseline to week 12 across renal function subgroups, except for patients with moderate renal impairment (normal renal function, p < 0.001; mild renal impairment, p < 0.001; moderate renal impairment, p = 0.572; Figure 5(a)). Additionally, significantly more prucalopride-treated than placebo-treated patients had a reduction in the time to first CSBM across all renal function subgroups (normal renal function, p < 0.001; mild renal impairment, p < 0.001; moderate renal impairment, p = 0.043; Figure 5(b)). Other secondary efficacy endpoints in patients stratified by renal function are summarized in Table 6; for almost all of these efficacy endpoints, patients experienced greater improvements from baseline to weeks 1–12 with prucalopride than with placebo. The global severity of constipation and efficacy of treatment scores improved in prucalopride-treated patients compared with placebo-treated patients across all renal function subgroups at week 12 of treatment, apart from the global efficacy of treatment score in patients with moderate renal impairment (Supplemental Table S3).
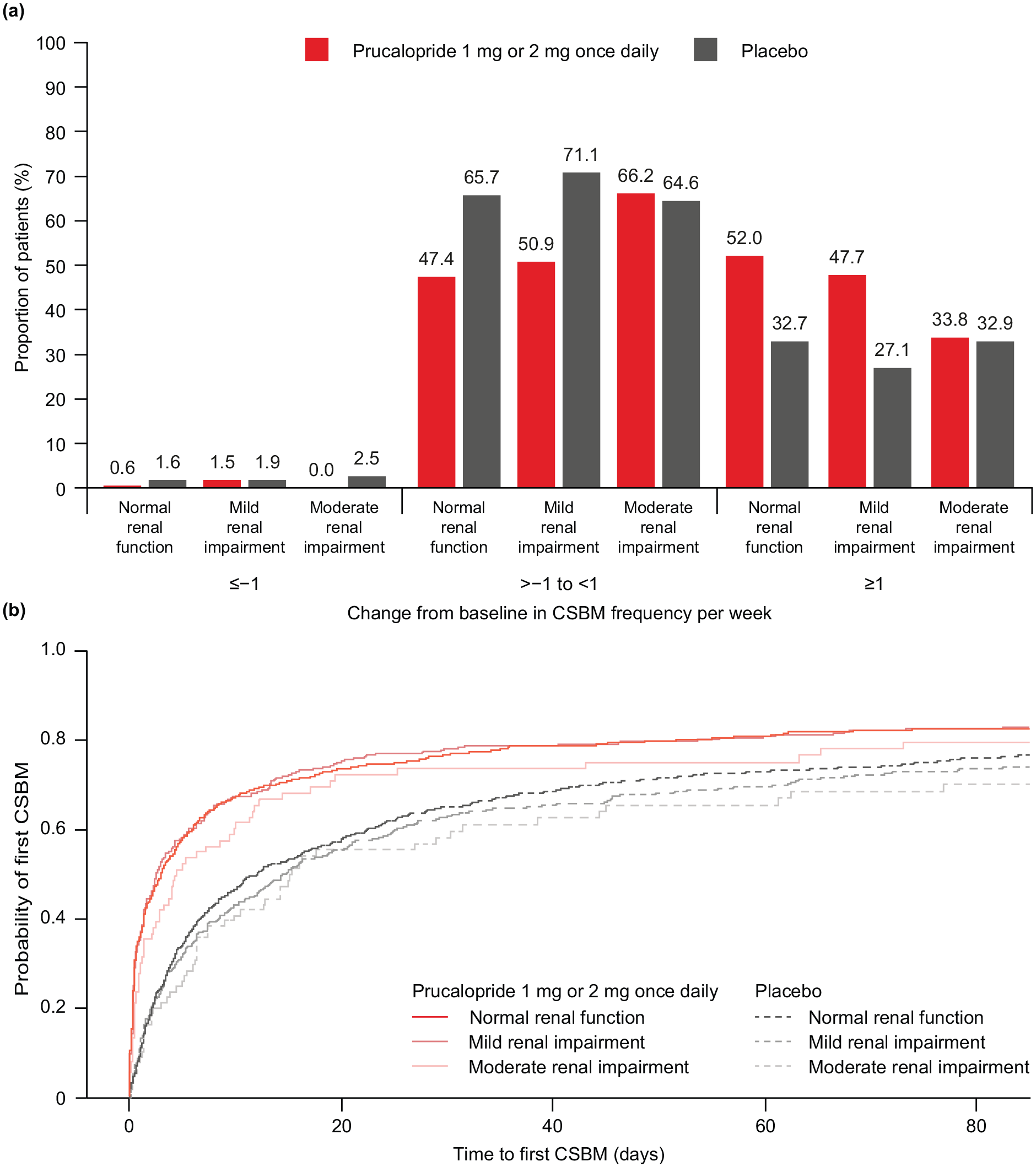
Change from baseline to week 12 in CSBM frequency per week in prucalopride-treated and placebo-treated patients (a),a and the time to first CSBM after the first dose of prucalopride or placebo (b),b stratified by renal function. Normal renal function, eGFR ⩾90 mL/min/1.73 m2; mild renal impairment, eGFR 60 to <90 mL/min/1.73 m2; moderate renal impairment, eGFR 30 to <60 mL/min/1.73 m2.
Summary of the secondary efficacy endpoints in prucalopride-treated and placebo-treated patients, stratified by renal function.

Normal renal function, eGFR ⩾90 mL/min/1.73 m2; mild renal impairment, eGFR 60 to <90 mL/min/1.73 m2; moderate renal impairment, eGFR 30 to <60 mL/min/1.73 m2.
CI, confidence interval; eGFR, estimated glomerular filtration rate; LS, least-squares; PAC-QOL, Patient Assessment of Constipation Quality of Life questionnaire; PAC-SYM, Patient Assessment of Constipation Symptoms questionnaire; SD, standard deviation; SE, standard error.
The Supplemental Material provides further information on the findings for the other secondary efficacy endpoints including the proportion of stools with a normal consistency or a hard to very hard consistency, the proportion of bowel movements with no straining or with severe or very severe straining, rescue medication use, and PAC-SYM and PAC-QoL total scores in each subgroup.
Safety endpoints
The proportions of patients with any TEAEs were generally higher with prucalopride than with placebo across all age subgroups (Table 7). The proportions of prucalopride-treated patients with treatment-related TEAEs were similar in patients aged younger than 50 years (39.7%) and in patients aged 50–64 years (37.6%), but lower in those aged 65 years or older (21.6%). Similarly, the proportion of patients with severe TEAEs was highest in prucalopride-treated patients aged younger than 50 years (14.1%) and lowest in those aged 65 years or older (5.4%). The proportion of patients with TEAEs leading to study drug withdrawal was higher in prucalopride-treated patients aged younger than 50 years (4.4%) and those aged 50–64 years (7.5%) than in those aged 65 years or older (3.9%).
Summary of TEAEs and cardiovascular events of interest in prucalopride-treated and placebo-treated patients, stratified by age.

n is the number of patients experiencing the event and were counted once per category, irrespective of the number of events.
TEAE, treatment-emergent adverse event.
The proportions of patients who experienced any TEAEs were higher with prucalopride than placebo across the underweight/healthy weight, overweight, and obese subgroups (Table 8). The proportions of prucalopride-treated patients with treatment-related TEAEs were similar in underweight/healthy weight (39.5%), overweight (31.9%), and obese (32.8%) patients. The proportion of patients with severe TEAEs was highest in prucalopride-treated patients in the underweight/healthy weight (13.8%) and overweight (11.4%) subgroups and lowest in obese patients (6.1%). The proportions of patients with TEAEs leading to study drug withdrawal were higher in prucalopride-treated patients in the underweight/healthy weight (5.7%) and obese (5.0%) subgroups than in the overweight subgroup (4.3%).
Summary of TEAEs and cardiovascular events of interest in prucalopride-treated and placebo-treated patients, stratified by BMI.
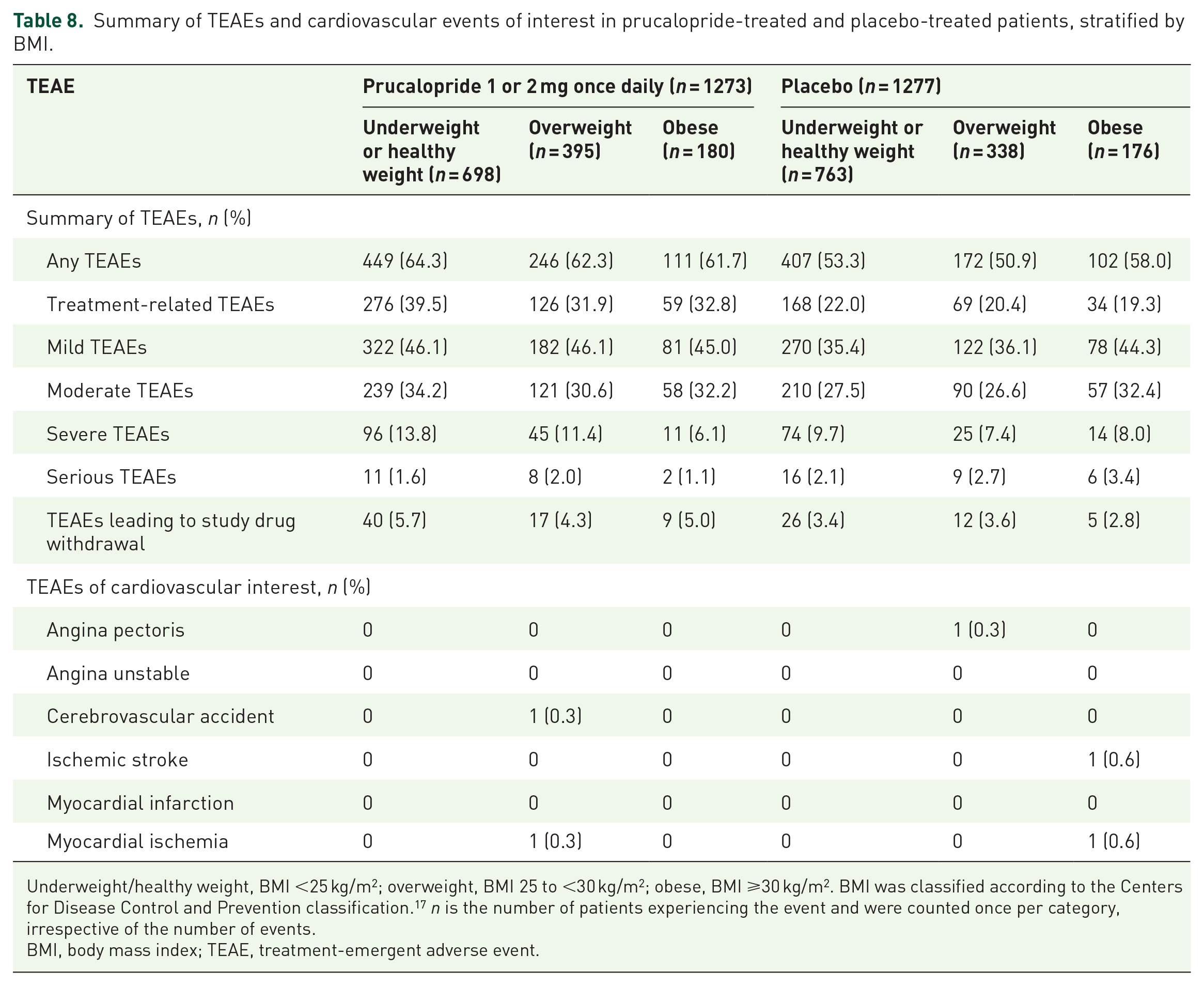
Underweight/healthy weight, BMI <25 kg/m2; overweight, BMI 25 to <30 kg/m2; obese, BMI ⩾30 kg/m2. BMI was classified according to the Centers for Disease Control and Prevention classification. 17 n is the number of patients experiencing the event and were counted once per category, irrespective of the number of events.
BMI, body mass index; TEAE, treatment-emergent adverse event.
The proportions of patients with any TEAEs were higher with prucalopride than with placebo in the normal renal function and mild renal impairment subgroups but were similar for prucalopride and placebo in the moderate renal impairment subgroup (Table 9). The proportions of prucalopride-treated patients with treatment-related TEAEs were 35.0%, 39.5%, and 28.8% in the normal renal function, mild renal impairment, and moderate renal impairment subgroups, respectively. An increase in the proportion of patients with treatment-related TEAEs was observed with prucalopride compared with placebo. The proportions of prucalopride-treated patients with severe TEAEs were 11.0%, 14.1%, and 10.0% in the normal renal function, mild renal impairment, and moderate renal impairment subgroups, respectively. There was no clear difference in the proportion of patients with serious or severe TEAEs between the prucalopride and placebo treatment groups. The proportions of patients who had a TEAE leading to study drug withdrawal ranged from 4.5% to 7.5% in the prucalopride group and from 2.0% to 9.8% in the placebo group. The proportions of patients with a TEAE leading to study drug withdrawal were highest in those with moderate renal impairment.
Summary of TEAEs and cardiovascular events of interest in prucalopride-treated and placebo-treated patients, stratified by renal function.

Normal renal function, eGFR ⩾90 mL/min/1.73 m2; mild renal impairment, eGFR 60 to <90 mL/min/1.73 m2; moderate renal impairment, eGFR 30 to <60 mL/min/1.73 m2. n is the number of patients experiencing the event and were counted once per category, irrespective of the number of events.
eGFR, estimated glomerular filtration rate; TEAE, treatment-emergent adverse event.
TEAEs of CV interest were not observed in prucalopride-treated patients aged younger than 50 years (Table 7), in patients in the underweight/healthy weight or obese subgroups (Table 8), or in patients with normal renal function or moderate renal impairment (Table 9). Only two prucalopride-treated patients (2 mg dose), both in the overweight subgroup, reported TEAEs of CV interest (cerebrovascular accident and myocardial ischemia, both of which were considered serious AEs; Table 8). The event of cerebrovascular accident was reported in a patient aged 65 years or older with mild renal impairment, and the event of myocardial ischemia was reported in a patient aged 50–64 years (renal function not available). The cerebrovascular accident required hospitalization and was considered moderate in severity but unlikely to be related to the study drug; therefore, the patient continued to receive prucalopride. The event resolved and the patient withdrew consent and discontinued the study. The myocardial ischemia event was considered mild in severity and possibly related to the study drug. The study drug was withdrawn and the patient discontinued the study. No deaths occurred in the six clinical studies assessed. Full safety data for the individual studies are presented elsewhere.24–29
Discussion
With an aging population and increasing levels of obesity,10,31 there is a need to understand the effect of age and BMI on the efficacy and safety of medicines. Although CIC may disproportionately affect older patients and those with a higher BMI,3–5 the effects of age and BMI on the efficacy and safety of treatments for CIC, such as prucalopride, have not been well characterized.4,5 Moreover, prucalopride is predominantly excreted by the kidneys,12,14 and dose reductions are recommended for patients with severe renal impairment. 11 Understanding how age, BMI, and renal function affect the benefit–risk profile of prucalopride is therefore important to help clinicians to determine whether any special considerations are needed when treating patients with CIC using prucalopride.
This post hoc analysis of six phase III–IV clinical trials found that a significantly greater proportion of patients with CIC treated with prucalopride 1 or 2 mg once daily than placebo-treated patients achieved a mean frequency of at least three CSBMs per week over 12 weeks of treatment, irrespective of their age. The proportion of prucalopride-treated patients who experienced an increased mean frequency of at least three CSBMs per week over the 12-week treatment period was higher in patients with a low BMI than in those with a higher BMI, and in patients with normal renal function or mild renal impairment than in those with moderate renal impairment. These findings suggest slight differences in the efficacy profile of prucalopride in patients with CIC by BMI and renal function. Except for patients who were classified as overweight and those who had moderate renal impairment, significant improvements were also observed in prucalopride-treated patients across all subgroups for several secondary efficacy endpoints, including an increase in the CSBM frequency per week from baseline and a faster time to first CSBM, compared with placebo. Significant improvements were not observed in prucalopride-treated patients who were obese or who had moderately impaired renal function for primary and secondary efficacy endpoints compared with placebo-treated patients. Lastly, prucalopride was well tolerated across all subgroups, including in patients aged 65 years or older, with no unexpected safety concerns identified.
Diet and lifestyle changes are often ineffective for managing CIC in older adults. 32 Although over-the-counter laxatives may alleviate symptoms in some patients with CIC and nonmodifiable risk factors, other agents may be needed to treat laxative-resistant constipation. 33 Two studies to date have investigated the efficacy and safety of prucalopride for the treatment of CIC in patients aged 65 years or older. In a double-blind, placebo-controlled study of 300 patients with constipation who were randomized to receive prucalopride (1, 2, or 4 mg once daily) or placebo for 4 weeks, a higher proportion of prucalopride-treated patients achieved at least three CSBMs than placebo-treated patients over 4 weeks of treatment. 34 The proportions of treatment-related TEAEs were similar in the prucalopride and placebo groups. In another double-blind, placebo-controlled study that investigated the safety of prucalopride (0.5, 1, or 2 mg once daily) for 28 days in 89 nursing home residents aged 65 years or older with constipation, 35 prucalopride was well tolerated. Additionally, no CV safety concerns were identified in this patient population, which had a high incidence of baseline CV disease. 35 These data are similar to the findings of the post hoc analysis reported here.
The effect of body weight on the efficacy and safety of prucalopride has not been investigated previously. Although our analysis did not show a significant increase in the proportion of prucalopride-treated patients achieving the primary efficacy endpoint compared with placebo-treated patients in the obese subgroup, the sample size for this subgroup was smaller than for the underweight/healthy weight and overweight subgroups. A previous analysis of prucalopride found no clinically significant differences in its pharmacokinetic profile based on body weight (after accounting for the effect of renal function). 11 However, patients who are obese may experience underdosing, which may lead to reduced treatment efficacy. 9 Conversely, although limited evidence is available on how the pharmacokinetic profile of a drug varies in underweight individuals, there may be an increased risk of AEs owing to a potential higher drug concentration in the blood.36–38 It is not clear why underweight patients experienced more AEs than patients in other weight subgroups; further studies are required to understand the effect of weight and BMI, particularly at the lower and higher ranges. It should also be noted that during our study, underweight patients were not examined separately, owing to the small sample size, and were combined with patients of a healthy weight. Similarly, it has been reported that impaired renal function can alter the pharmacokinetic properties of a drug, which can lead to reduced drug efficacy or an increased risk of adverse effects.39,40 The reduced therapeutic effect observed in patients with moderate renal impairment in our study may be due to the small sample size compared to the normal renal function and mild renal impairment subgroups. Additional research in this subgroup is required to understand whether the efficacy of prucalopride is altered in patients with moderately impaired renal function.
There was no clear relationship between the incidence or nature of TEAEs and age, BMI, or renal function in patients receiving prucalopride. Prucalopride was well tolerated, and no unexpected safety concerns were identified. Although the previously available serotonin type 4 receptor agonists cisapride and tegaserod have been shown to be nonselective and associated with adverse CV events, 41 studies to date have not raised concerns regarding the impact of prucalopride on CV safety. 42 CV events of interest were not observed in prucalopride-treated patients for most age, BMI, and renal subgroups in this post hoc analysis. Two serious CV events of interest were reported in two prucalopride-treated patients in the overweight subgroup. One patient, who also had mild renal impairment, experienced a moderate event of cerebrovascular accident that was considered unlikely to be related to the study drug, and one patient experienced a mild event of myocardial ischemia that was considered to be possibly related to the study drug. This is the first post hoc analysis to evaluate safety endpoints in patients with renal impairment who are receiving a serotonin type 4 receptor agonist for the treatment of CIC.
The efficacy of prucalopride 1 mg has been demonstrated in patients with CIC and severe renal impairment in the USA and in patients with CIC aged 65 years and older in Europe, and is the approved dose in these populations.11,13,23 During the studies included in this post hoc analysis, only 21 patients received prucalopride 1 mg once daily, whereas 1216 patients in the age and BMI subgroups and 1212 patients in the renal function subgroups received prucalopride 2 mg once daily. Therefore, we do not anticipate that these subpopulations will have a substantial impact on the overall efficacy and safety endpoints reported here.
One limitation of our analysis is that it was performed post hoc, and such analyses are not generally powered to show statistical differences. The analysis was also limited by the small proportion of patients who were obese, were aged 65 years or older, or had moderate renal impairment; these subgroups represented only 13.9%, 15.1%, and 6.5% of the total population, respectively. Further investigations in these subgroups would confirm the findings from our analyses. In addition, most of the patients included in our post hoc analysis were White. Including more diverse patient populations (e.g., a higher number of male patients, older patients, and patients from different racial and ethnic groups) in future clinical studies would help to improve understanding of the efficacy and safety of prucalopride for the treatment of CIC and would provide valuable insights into the impact of treatment in a patient population that more closely resembles a real-world population of patients with CIC.
Although this was a post hoc analysis, it combined data from six key phase III and IV clinical studies with prespecified endpoints,24–29 resulting in a large overall sample size of patients. Furthermore, this is the first efficacy and safety analysis to stratify patients with CIC by either age, BMI, or renal function who were receiving prucalopride 1 or 2 mg once daily compared with placebo. This is also the first analysis to specifically investigate the impact of age, BMI, and renal function on the CV safety profile of prucalopride.
Conclusion
This post hoc analysis showed that prucalopride is efficacious in adult patients with CIC of different ages (<50, 50–64, and ⩾65 years), in those who are underweight/healthy weight or overweight, and in those with normal renal function or mild renal impairment. Prucalopride-treated patients who were obese or who had moderate renal impairment also had improvements in the primary efficacy endpoint compared with placebo-treated patients, but these findings were not statistically significant. Prucalopride 1 or 2 mg once daily was well tolerated, with most AEs being mild to moderate in severity and unrelated to the study drug. Further studies in a more diverse patient population are required to improve our understanding of the impact of prucalopride treatment across the CIC population spectrum.
Supplemental Material
sj-docx-1-tag-10.1177_17562848241299731 – Supplemental material for Efficacy and safety of prucalopride in patients with chronic idiopathic constipation stratified by age, body mass index, and renal function: a post hoc analysis of phase III and IV, randomized, placebo-controlled clinical studies
Supplemental material, sj-docx-1-tag-10.1177_17562848241299731 for Efficacy and safety of prucalopride in patients with chronic idiopathic constipation stratified by age, body mass index, and renal function: a post hoc analysis of phase III and IV, randomized, placebo-controlled clinical studies by Anthony Lembo, Kyle Staller, Mena Boules, Paul Feuerstadt, William Spalding, André Gabriel, Ashraf Youssef, Yunlong Xie, Brian Terreri and Brooks D. Cash in Therapeutic Advances in Gastroenterology
Footnotes
Acknowledgements
The authors thank Yaping Wan for their valuable contributions to these analyses. Medical writing support was provided by Tsvetana Stoilova, PhD, Sandra Cheriyamkunnel, MSc, and Joanna L. Donnelly, PhD, of PharmaGenesis London, London, UK, and was funded by Takeda Pharmaceuticals USA, Inc.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.