
Abstract
The gut microbiome comprised of microbes from multiple kingdoms, including bacteria, fungi, and viruses. Emerging evidence suggests that the intestinal fungi (the gut “mycobiome”) play an important role in host immunity and inflammation. Advances in next generation sequencing methods to study the fungi in fecal samples and mucosa tissues have expanded our understanding of gut fungi in intestinal homeostasis and systemic immunity in health and their contribution to different human diseases. In this review, the current status of gut mycobiome in health, early life, and different diseases including inflammatory bowel disease, colorectal cancer, and metabolic diseases were summarized.
Introduction
Most studies of the human gut microbiome have focused on the bacterial component of the microbiome, but the fungal microbiome (i.e. the mycobiome) has recently gained recognition as a fundamental part of the human microbiome. Fungi inhabit the mucosal surface to maintain intestinal homeostasis and systemic immunity. 1 Until recently, there has been a lack of research focusing on the interactions of the fungal kingdom of microorganisms with other constituents of the gut microbiome and their contribution to health and diseases. To date, less than 3% of the microbiome literature accounts for the presence of fungi in microbial communities. Recent advances in sequencing technology have provided comprehensive tools to profile the fungal component of the gut microbiome 2 and emerging data suggest that the gut mycobiome may act as a reservoir for potentially opportunistic pathogens in inflammatory bowel disease (IBD),3,4 graft-versus-host disease (GVHD), 5 gastrointestinal cancer,6–8 and other diseases. Here, we discuss the importance of developing standardized methodology to define the gut mycobiome in early life, health, and diseases. We also highlight future directions of studies to delineate the role of gut mycobiome and the potential of therapeutic intervention targeting the gut mycobiome.
Methods to characterize the gut mycobiome
Characterization of the gut mycobiome has been complicated by the lack of comprehensive, well-curated, and high-resolution taxonomic annotation within fungal databases. Development of standardized methods for mycobiome analyses is critical to minimize variations across studies. A comprehensive literature search using broad searching criteria was conducted in the PubMed and Google Scholar databases. In short, 103 relevant literatures related to gut mycobiome are carefully selected based on the key words including “gut mycobiome”, “fungi”, “fungus”, “eukaryotes”, “fungal community”, “yeast,” and “sequencing”. The fungal study workflow contains extraction, library construction, and sequencing, and bioinformatic analysis was shown in Figure 1(a). There were 33 different commercial extraction kits used for fungi studies2,5,6,8–74 with 13 customized protocols.5,9,21,26,34,35,39,47,53,58,73–75 QIAamp DNA Mini Kit (Qiagen) was the most commonly used kit in 16 papers.2,6,8,16,22,23,33,43–47,49,54,61,67 Fast DNA Spin Kit (MP Biomedicals) was a common extraction kit for fungi.11,12,17,27,36,65,66,70
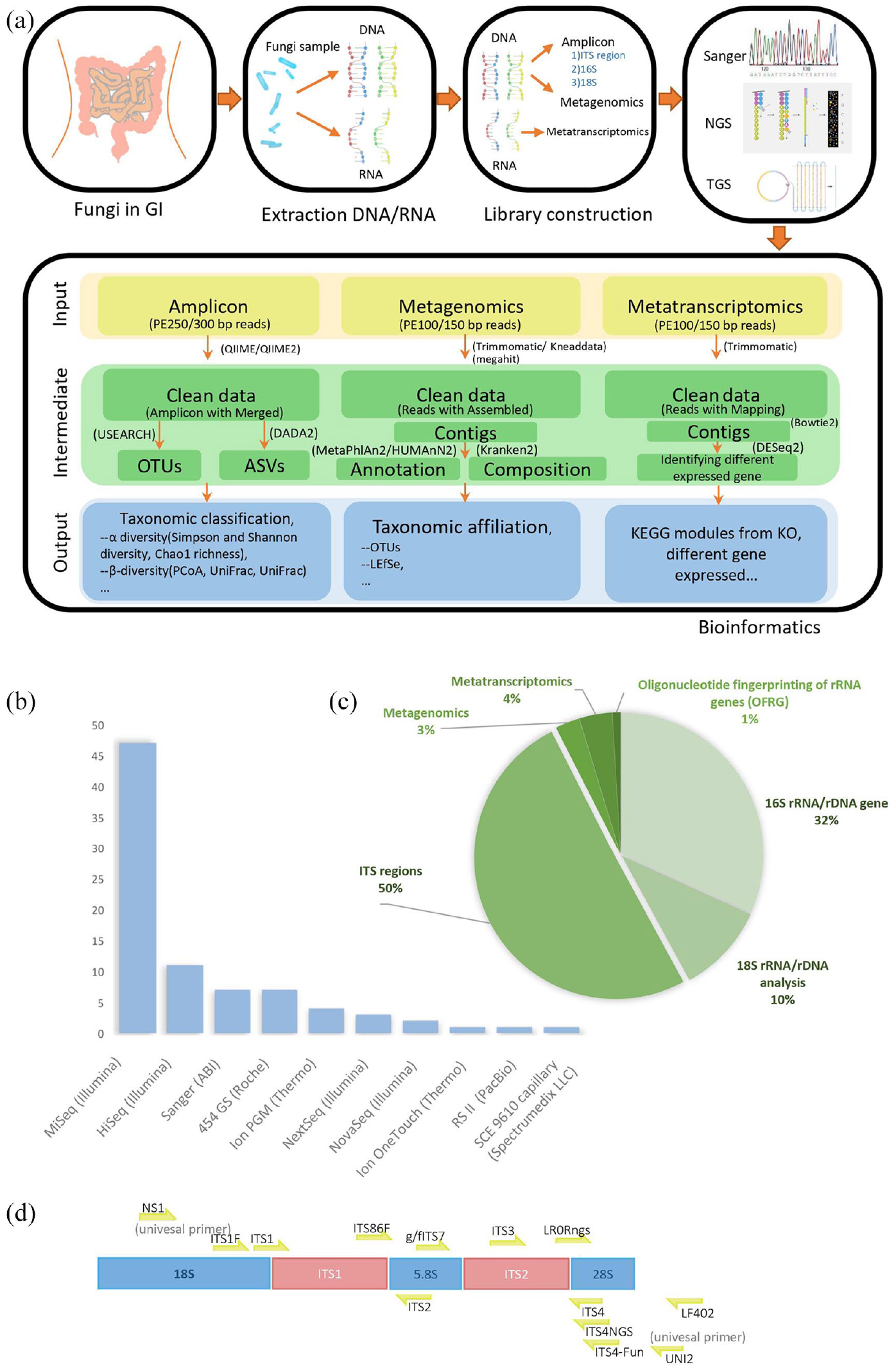
The fungal study workflow contains extraction, libraries construction, sequencing, and bioinformatic analysis. (a) Initially DNA/RNA will be isolated from fungal samples. Then DNA or RNA extraction was performed following the protocol of amplicon, metagenomics, and metatranscriptomics according to strategy. Depending on strategy of research, different sequencer is selected from first-generation sequencing to third-generation sequencing. Generally, next generation sequencing (NGS) platform is chosen by higher output like Illumina sequencing by synthesis (SBS) sequencing technology, while-third generation sequencing has also become competitive for sequence full-length small genomes without assembly like Pacbio Single-molecule real-time (SMRT) sequencing. Analyze sequencing data using amplicon, metagenomics, and metatranscriptomics bioinformatics pipeline to analysis 16S rRNA/rDNA, DNA and RNA, respectively. (b) Among the studies involving sequencing methods, there are 10 sequencing platforms are used, in which NGS platform is used frequently and Illumina MiSeq platform is used the most in NGS method. (c) The fungal sequencing strategies are the study of ITS region, analysis of 16S rRNA/rDNA and 18S rRNA/rDNA gene. (d) ITS sequences and commonly and locatable primers used in fungal target region.
About 90% of users selected next generation sequencing (NGS) platform for higher throughput and faster turnaround time. Among all the sequencing platforms, Illumina MiSeq platform is the most widely utilized sequencer for 16S amplicon (Figure 1(b)). Widely used platform of NGS is Illumina which has high throughput, high quality, and high speed for large number of samples. Compared with Nanopore which belongs to one of the third-generation sequencing (TGS) technique, Illumina sequencers involve the following features: (1) it has implemented hundreds of metabarcoding of complex samples per run through dual-index method compared with less than 24 sequences in a Nanopore single run. 76 (2) It has high-quality rate with 99.99% accuracy higher than approximately 90% of Nanopore. 77 (3) For each sequencing run of massive fungal specimen, the MiSeq generates 24Gbp with PE300 and the NovaSeq 6000 generates 2190Gbp with PE150, respectively, but 10Gbp was generated by MinION in general research lab. 78 With the continuous development of sequencing technology, new technologies have been used in research to identify fungi, especially Nanopore, 79 and TGS sequencers have two significant advantages compared with Illumina. Initially, as an advantage of long reads sequencing in Nanopore, 0–200 kbp DNA could be covered, while 400–600 bp reads are sequenced in illumina sequencing. Long fragment could be reconstructed and identified as species precisely, decreasing the number of mislabeled species identified in illumine. 80 Secondarily, researches use Nanopore to detect fungi for the advantage of amplicon-based and PCR-free metagenomics technology. 77
For sequencing strategy, as indicated in Figure 1(c), 50% of articles sequenced the internal transcribed spacer (ITS) region, including ITS1 and ITS2, while 32% articles focused on 16s rRNA/rDNA gene and 10% papers on 18s rRNA/rDNA regions. Only 3% of the papers have performed metagenomics and 4% with metatranscriptomic fungal analysis. The fungal ribosomal region contains the internal transcribed spacers (ITS1 and ITS2) and the 5.8s, 18s, and 28s rRNA (Figure 1(d)). Currently, ITS1 and ITS211–16,51,58,65,66,73,75,81 are two major optional primers for human gut mycobiome study and the primer ITS2 is longer than primer ITS1 which has lower species resolution but fewer amplification and sequencing errors. 82 Besides, the primer ITS1 is biased toward the amplification of Basidiomycetes and the primer ITS2 for the Ascomycetes. 83 There were also reported ITS target regions outperformed compared with other subunit rRNA region in the human microbiome. 28 Because ITS regions show not only fewer number of ribosomal gene copies but also less relative abundance of variants and breakpoints in various regions per unit in human rDNA. 84 A preview results nonetheless support that the result of 18s rDNA PCR, which compares with one of ITS PCR, shows significant diagnostic and accurate identification of fungal pathogens in clinical sample from fresh biopsies and punctate and deep wound secretions. 85 Compared with target sequence, metagenomics reads might be decreased biases from primer choices and increased community taxonomic characterization providing information of gene composition and the function. 86
Overall, although significant advancements have been made on bioinformatics methodologies for gut mycobiome analyses, further development of pipelines and databases is required to define and characterize fungal communities more accurately.
The gastrointestinal tract mycobiome
Fecal samples are the most commonly used specimens to represent the human gut mycobiome. According to Human Microbiome Project, Saccharomyces, Malassezia, Candida, Cyberlindnera, Penicillium, Cladosporium, Aspergillus, Debaryomyces, Pichia, Clavispora, and Galactomyces are the most prevalent fungal genera in the human gut based on ITS2 and 18S rRNA sequencing. 87 Cultivable gut mycobiota including Candida albicans, Candida glabrata, Candida deformans, Aspergillus glaucus, Cryptococcus saitoi, Cryptococcus neoformans, Lichtheimia ramosa, Mucor circinelloides, Pleurostomophora richardsiae, Rhodotorula mucilaginosa, Trichosporon asahii and Yarrowia lipolytica are frequently isolated from human feces.88,89 By pyrosequencing, Hoffmann et al. 90 have found 12 fungal genera in fecal samples and Saccharomyces (present in 89% of the samples) was the most dominant. In another study of 45 healthy individuals, a total of 72 distinct operational taxonomic units (OTU) of gut fungi were found using ITS sequencing. Candida tropicalis, Geotrichum gigas, C. sake, and Phichia jadinii were the most commonly detected fungal microbiota from the fecal samples. 91 Moreover, edible mushrooms (Agaricus bisporus) and plant pathogen (Epicoccum nigrum and Alternaria spp.) were also identified in the gastrointestinal tract of vegetarian individuals. 43 Fecal-associated mycobiome are less persistent and varied over time and is easily influenced by food intake with “passing through” fungi.91,92 For instance, Saccharomyces cerevisiae and C. albicans were the two abundant fungal species in healthy individuals’ gut.92,93 However, in a controlled-diet experiment, Saccharomyces declined below the limit of detection in stool when the volunteer consumed a S. cerevisiae-free diet, and the levels of C. albicans in stool were dramatically reduced when the volunteer cleaned their teeth more frequently. 71
Mucosa is also colonized by fungi. Compared with luminal-associated gut mycobiota, a more stable fungal community was found in the intestinal mucosa.6,94 There is limited research reporting the fungal composition of mucosa tissues as specimens are less accessible compared with fecal samples. 95 A study applied 18s rDNA sequencing to study the mucosa-associated fungi of colonic biopsy tissues from 47 healthy individuals. They found that R. mucilaginosa, Galactomyces geotrichum, C. albicans, Septoria epambrosiae, Cryptococcus carnescens, Bullera crocea, C. dubliensi, Cladosporium cladospoirioides, Raciborskiomyces longisetosum, and Penicillium ialicum were the most detected in mucosal samples. 96 Future studies comparing fecal and mucosa mycobiota and the contribution of their individual role in intestinal homeostasis and pathogenesis of disease are warranted.
Factors affecting the gut mycobiome
Despite emerging research on the gut mycobiome, a consensus healthy mycobiome has yet to be established. Several factors have been shown to be associated with alterations in mycobiota community composition including host genetics, gender, age, comorbidities, drugs, lifestyle factors including hygiene, socioeconomic status, diet, occupation, and the immune system. 81 Interestingly, unlike the bacteriome, the mycobiome was shown a higher diversity in highly acidic stomach environment in piglet. 97 This suggested that the imbalance of gut mycobiota may be related to stomach disease. Indeed, recent study reveals a perturbation of fungal compositional and ecological changes in gastric cancer development and C. albicans was characterized as a biomarker for gastric cancer. 98 Given that early life factors are known to influence host microbiome status, data on early life gut mycobiome and factors influencing their development were reviewed.
Early life gut mycobiota
Early life fungal colonization has an impact on health outcomes of infants by training their immune system. Gut mycobiome in early life and factors that influence their development are shown in Figure 2. Infant fecal mycobiome was characterized by a low relative abundance of fungi composition and richness. Mother and infant fecal mycobiome were profiled in 15 mother–infant pairs in the first year of life by 18s rRNA gene amplicon sequencing. 99 Phyla Basidiomycota and Ascomycota, with the genus Saccharomyces and the class Exobasidiomycetes, were the most represented fungal component found in meconium. In another study, 100 fecal samples from 298 mother–offspring pairs were analyzed by ITS1 amplicon sequencing. S. cerevisiae was the most abundant species in the gut of infants from 1 year of age onwards. Besides, Debaryomyces hansenii prevailed up to 3 months in the infant’s feces. The 10-day, 1-year, and 2-year fecal samples from infants were richer in R. mucilaginosa, whereas the 3-month samples showed a high colonizer of C. parapsilosis. To further investigate early fungal community establishment, Ward et al. 101 assessed the skin, oral, and anal mycobiomes of 16 infants over the first month of life and the anal and vaginal mycobiomes of 17 mothers using ITS2 amplicon sequencing. Infant mycobiomes varied by three body sites; skin mycobiome were dominated by C. tropicalis, C. parapsilosis, S. cerevisiae, C. albicans, and C. orthopsilosis; oral mycobiome was enriched with C. parapsilosis, C. tropicalis, S. cerevisiae, C. orthopsilosis, C. albicans, and Cladosporium velox; and anal mycobiome were colonized with C. parapsilosis, C. tropicalis, C. albicans, S. cerevisiae, C. orthopsilosis, and Cryptococcus pseudolongus, respectively. 99 However, the infant’s mycobiome did not show a trajectory toward maturity within first 30 days of life, and this could be in part explained by the use of a relatively consistent food source of either breast milk or infant formula. Moreover, Kasper has demonstrated that mycobiota could be transferred from mothers to their offspring. In the study, five infants shared D. hansenii and S. cerevisiae observed in their mothers. 100 Notably, the mother-to-child bacterial transmission patterns have been well profiled at strain level,102,103 whereas limited studies on mother-to-child vertical fungal transmission were reported. Overall, these studies all demonstrated that fungi could colonize in the neonatal gut at very early stage; nevertheless, how the mycobiome is shaped and how the succession occurs in the gut of neonates remained to be elucidated in the future.

The gut mycobiome through life and the influencing factors in early life. Infants receive mycobiota colonization in the gut at birth, and fungal community increased with age in infancy but decreased when they grow up to young adults. 10 days after birth, Rhodotorula mucilaginosa and Debaryomyces hansenii predominated in the gut of infants, while Candida parapsilosis, C. tropicalis, C. albicans, Saccharomyces cerevisiae, C. orthopsilosis, and Cryptococcus pseudolongus enriched in the anus of infants in the first month. In the adult stage, C. albicans, S. cerevisiae, C. tropicalis, C. glabrata, C. deforans, and Aspergillus glaucus occupy the gut. In later years, Penicillium, Candida, Aspergillus, and Saccharomyces were dominant in the gut of elders. The major factors contributing to neonatal gut fungal communities were mode of feeding and mode of delivery. Compared with neonates by cesarean section, a higher level of Candida and Pleosporales, decreased level of Malassezia were shown in the gut of infants born via vaginal delivery. Moreover, infants could directly acquire mycobiota from breast milk feeding by their mothers, which contains abundant R. mucilaginosa and C. parapsilosis.
Factors affecting mycobiome in early life
Fungi are ubiquitous in the environment and the infant mycobiome may originate from the mother during birth, from the mother’s breast milk, parental skin, or anywhere else in the hospital or home environment with which the offspring come in contact with. 100
Mode of delivery. In newborn babies, initial fungal colonization is largely dependent on delivery mode (Figure 2). Infants born vaginally obtained fungi that colonize the vagina, whereas infants born via Caesarian section acquire fungal species that are related to the skin.104–106 Azevedo et al. 107 found that delivery mode might be associated with a higher carriage of oral fungi at a young age. Ward et al. 108 highlighted that the mode of birth likely influence the fungal composition of the infant. It was believed that infants born vaginally appeared to have a higher level of Candida in their fecal mycobiome given that Candida was found predominantly in the vagina canal and they also had a more diverse mycobiome compared to those born by cesarean section (C-section) given the exposure of the infant to the mother’s fecal mycobiota. In contrast, infants delivered by C-section were colonized by a relatively higher abundance of Malassezia which originated from their mother’s skin. However, there are limited studies to support these hypotheses. In a separate study, Zhu et al. 109 found that relative abundance of the order Pleosporales was higher in infants born by vaginal delivery than in those born by C-section.
Mode of feeding. Breast milk is considered the most ideal nutrition for infants offering protection from neonatal sepsis and facilitating infant growth and development. 110 Several studies have shown that the mode of feeding impact on the gut bacterial microbiome.111,112 Boix-Amorós et al. 113 characterized mycobiome composition in breast milk from healthy mothers and showed that the fungal composition of human breast milk was dominated by Malassezia (44%), followed by Candida (19%) and Saccharomyces (12%). The most abundant viable fungi detected were R. mucilaginosa and C. parapsilosis (Figure 2). 24 Azevedo et al. 107 suggested that the feeding mode of the infant (formula-fed or breast-fed infants) did not result in a difference in their oral yeast carriage. In contrast, Ward et al. 108 suggested that feeding mode might affect the infant oral mycobiota. Currently, the relationship between infant and maternal gut mycobiome and whether breast milk leads to transmission of mycobiome from mother to infant remain controversial and require future studies. Future studies should focus on longitudinal tracking of the early-life mycobiota using mother–baby pairs while monitoring health outcomes during the first year of life. In addition, given the known interactions between the bacteria and fungi, future studies should also focus on the analysis of both bacteria and fungi to better characterize their structural and functional relationships, as well as their combined effects on health outcomes.
Gut mycobiota in elder subjects
Fungal community increased with age in infancy but decreased when they grow up to young adults (Figure 2). 88 However, gut mycobiota alteration in elder subjects was less reported, 114 and most of the older adults studied were reported with known diseases including hypertriglyceridemia (HG), 115 Alzheimer’s disease (AD),51,116 and type 2 diabetes mellitus (T2DM). 19 In Denmark, gut mycobiome analysis was performed in 100 elderly participants (70 with normotriglyceridemia versus 30 with hypertriglyceridemia) aged 65–81 years by ITS 2 amplicon sequencing on their fecal samples. Penicillium, Candida, and Aspergillus were the top three genera among the elderly Danes, and genus Penicillium was strongly correlated with the hypertriglyceridemia. 115 Another study on 17 older subjects (11 mild cognitive impairment versus 6 cognitively normal) with average age of 64.6 years showed that Saccharomyces, Candida, and Aspergillus were major fungal genera in their fecal samples through ITS 1 sequencing. Higher abundance of Botrytis, Kazachstania, Phaeoacremonium, and Cladosporium and decreased abundance of Meyerozyma at genus level were found in the gut of patients with mild cognitive impairment compared with controls. 51 Besides, Alonso et al. 116 suggest that higher proportion of fungi were found in the brain tissue of older patients with AD compared with controls, and the percentages of Aspergillus and Candida were higher in elder controls than that in young controls by ITS 1 amplicon sequencing. However, to date no studies have fully profiled the longitudinal changes of mycobiota in the gut of old adults. Further studies are needed for a more complete picture of microbial development throughout the whole life.
The role of gut mycobiome and disease susceptibilities
Mycobiota and inflammatory bowel disease (IBD)
IBD, including ulcerative colitis (UC) and Crohn’s disease (CD), is a chronic and relapsing, inflammatory disorder of the gastrointestinal tract. Its pathogenesis involves a complex interaction among host genetics, host immunity, microbiome, and environmental exposures. 117 The alterations of fecal and mucosal mycobiota in IBD in the pediatric and adult populations were summarized in Figure 3. As early as 1990, anti-Saccharomyces cerevisiae antibodies (ASCA) were found to be significantly higher in sera of patients with CD, which suggests that the gut mycobiota may be involved in the pathogenesis of CD. 118 Several studies have also confirmed increased ASCA titers in sera of CD patients and considered it as a potential biomarker for disease diagnosis.119–121 C. albicans was reported to be one of the immunogens for ASCA. 122 Standaert-Vitse et al. 122 showed that the pathogenic C. albicans in human tissues may induce overexpression of ASCA major epitopes. It was also shown that patients with CD and their first-degree healthy relatives were more frequently and more heavily colonized by C. albicans than healthy controls based on traditional culture method. 123 Moreover, in vivo experiments suggested that colonic inflammation facilitated C. albicans colonization in dextran sodium sulfate-induced mice colitis model.124,125

Schematic summary of the gut mycobiome alterations in IBD. Compared with healthy individuals, the gut mycobiome of patients with IBD is characterized by increased fungal diversity and mycobiota dysbiosis accompanying with inflammation. Increased Basidiomycota/Ascomycota ratio and Candida albicans level, decreased proportion of Saccharomyces cerevisiae were shown in the fecal samples of patients with IBD. Similarly, increased fungus load including of C. albicans, C. tropicalis, C. glabrata, and Gibberella moniliformis were also found in the intestinal mucosa of patients with IBD. Nevertheless, pediatric patients with IBD showed a decreased fungal diversity in the gut. C. albicans, C. utilis, C. parapsilosis, S. cerevisiae, Clavispora lusitaniae, and Kluyveromyces marxianus prevailed in the feces of children, while increased abundance of Psathyrellaceae, Cortinariaceae, Psathyrella, and Gymnopilus were detected in their mucosa.
Based on denaturing gradient gel electrophoresis (DGGE) analysis, Ott et al. 96 showed that fungal diversity was increased in patients with CD compared with controls. Sokol et al. 126 reported a distinct fungal microbiota dysbiosis in stool of patients with IBD characterized by an increased Basidiomycota/Ascomycota ratio, decreased proportion of S. cerevisiae, and an increased C. albicans using ITS2 sequencing. Liguori et al. profiled the fungal composition in colonic mucosa of patients with CD and healthy subjects and found an increase in fungus load during CD flare. In addition, Cystofilobasidiaceae family, Dioszegia genera, and C. glabrata species were overrepresented in CD while Leptosphaeria and Trichosporon genera were reduced compared with healthy controls. Similarly, a higher richness in fungal diversity in the inflamed mucosa of treatment-naïve UC patients was detected when compared with noninflamed mucosa (Figure 3). However, after 5-ASA treatment, the richness of diversity declined. 33 Furthermore, increased diversity of mucosal fungal microbiota was associated with expression of tumor necrosis factor (TNF)-α and interferon (IFN)-γ, which are key factors in the pathogenesis of IBD. 4 In contrast to findings in adults, a reduced diversity in fungal microbiota was reported in the stool of pediatric patients with CD and UC. 127 Besides, the represented fungal taxa detected in stool of pediatric patients with CD also differed from those of adults. Chehoud et al. 127 showed that C. utilis and Candida parapsilosis were more common in stool samples of children with IBD than healthy children via ITS1 Region Gene Sequencing. El Mouzan et al. 128 investigated fungal microbiota composition in treatment-naïve children with CD and showed that Psathyrellaceae, Cortinariaceae, Psathyrella, and Gymnopilus were significantly increased in the mucosa, while Cortinariaceae, Hymenochaete, and Gymnopilus were enriched in stool samples. In addition, Lewis et al. reported that S. cerevisiae, Clavispora lusitaniae, Candida utilis, C. albicans, and Kluyveromyces marxianus were present in abundance in stools from 86 pediatric subjects with CD compared to controls using shotgun metagenomic analysis (Figure 3). Pediatric and adult subjects with CD shared a small proportion of fungal taxa but there is also distinct differences in the fungal species. 129 These difference may be due to different clinical phenotypes, drugs, and gut bacterial community between adults and pediatric IBD subjects. Numerous studies have highlighted the interaction between fungi and bacteria in IBD. Several correlations between bacteria and fungi were observed in a pediatric cohort, which varied between the healthy controls and patients with CD. 129 Hoarau reported a significant interkingdom associations in bacteriome and mycobiome in 13 familial clusters, which included 6 bacterial-fungal genus and 13 species-level correlations. 69 In a DSS-induced mice colitis model, Sovran et al. 35 found that the beneficial effects of Saccharomyces boulardii and pathogenic effects of C. albicans on colitis severity could be eliminated through a broad-spectrum antibiotic cocktail treatment, including ampicillin, neomycin, metronidazole, and vancomycin. Overall, the gut fungal microbiota is altered in IBD but how fungi are involved in the occurrence and development of IBD remains unclear. Modulation of the fungal microbiota can be considered as a therapeutic approach for IBD as certain strains including S. boulardii and S. cerevisiae have shown therapeutic effects in human and murine IBD models. More studies on host–fungi interactions are necessary to enhance our understanding on how fungal microbiota interact with other constituents of the gut microbiota and the mechanisms of these relationships, in association with pathogenesis and development of IBD. Novel approaches, such as dietary interventions, probiotics, fecal microbiota transplantation (FMT), and antifungal metabolites to restore the dysbiotic states of intestinal mycobiota could be considered in the future; however, further studies are needed to assess their safety and efficacy.
Gut mycobiome in colorectal cancer (CRC)
Studies have shown that gut microbiota dysbiosis is associated with CRC. 130 Besides bacteriome and virome, involvement of gut mycobiota in colorectal carcinogenesis has been increasingly recognized recently. Luan et al. compared mucosa-adherent fungal microbiota of paired biopsy samples of adenomas with adjacent normal colon tissue and found that gut fungal diversity in adenomas was decreased compared with adjacent normal tissues. At the genus level, the opportunistic pathogens Phoma and Candida accounted for an average of 45% of the fungal microbiota. They also found an OTU, assigned to Spizellomycetales, to be significantly enriched in adenomas compared with adjacent samples while one OTU, assigned to Paraglomerales, was significantly enriched in nonadvanced adenomas compared with adjacent tissues. More importantly, Fusarium and Trichoderma genera, respectively, were significantly enriched in adjacent biopsy samples in advanced adenoma compared with samples of nonadvanced adenoma which may be useful for early diagnosis. 6 In a mycobiome study of 29 polyps, 74 CRC patients, and 28 healthy controls, fecal mycobiota composition differed among the three groups with a significant increase in the ratio of Ascomycota to Basidiomycota from control to CRC samples suggesting that changes of the gut mycobiota may be associated with progression of tumorigenesis. In addition, a higher fungal diversity was shown in late-stage CRC than in early-stage CRC. 5 An increased proportion of opportunistic fungi Trichosporon and Malassezia was detected as the major contributor in the progression CRC. 7 In a large multicenter study of 184 patients with CRC, 197 patients with adenoma and 204 control subjects, the ratio of Basidiomycota/Ascomycota was reported to be higher in CRC than in controls. 8 In addition, six fungal features were detected to be enriched in CRC compared with controls at the genus level, including Malassezia, Moniliophthtora, Rhodotorula, Acremonium, Thielaviopsis, and Pisolithus. 8 A small cohort comparing mycobiota in patients with colitis-associated cancer (CAC) and sporadic cancer showed no difference in the fungal microbiota composition. 131 Overall, fungal dysbiosis has been found in adenomas, CRC, or CAC. Understanding fungal diversity and abundance in CRC is crucial to help elucidate the potential contribution of fungal species in colorectal tumorigenesis and delineation of fecal fungal dysbiosis might shed light on new opportunities for utilizing fungal species as noninvasive diagnostic biomarkers for CRC or its precursor lesions.
Gut mycobiome in metabolic diseases
Animal and human studies support a role of gut fungi in metabolic disease.17,19,132 In a mouse model, Heisel et al. 133 investigated the effects of obesogenic diet on fungal composition by ITS2 sequencing and found that S. cerevisiae in the gut was significantly more abundant in lean mice than in mice fed with a high-fat diet. In an obese subject (body weight index = 48.9), a higher fecal fungal diversity was observed compared with healthy lean individuals. 134 Furthermore, by using parallel aerobic culture-dependent approach on 24 subjects and ITS-based sequencing on 52 subjects, impaired fungal communities were reported in the gut of obese individuals, characterized by an increased presence of the phylum Ascomycota, the class Saccharomycetes, and the families Dipodascaceae and Saccharomycetaceae as compared with nonobese individuals.17,135 These findings suggest that obese subjects had altered gut fungi composition compared with their lean counterparts.
Mycobiome dysbiosis has also been reported in the gut of patients with diabetes mellitus.136–139 A higher diversity in fecal fungal species was found to distinguish children with type 1 diabetes mellitus (T1DM) from healthy controls. 136 Moreover, a significantly increase in Candida colonization was found in the fecal samples of patients with type 1 and 2 diabetes compared with controls, which was verified by quantitative real-time PCR and medium cultures.137,138 In contrast, a separate study reported that C. albicans was significantly less prevalent in individuals with T1DM (62% of all strains identified) compared to control subjects (85% of all strains identified) based on medium cultures. Moreover, the fungal species isolated in this study were shown to be more resistant to antifungal drugs. 136 Overall, the role of Candida in the pathogenesis of T1DM requires further confirmation. Using fungal ITS1 metagenomic sequencing on fecal samples from 10 healthy controls, 14 newly diagnosed T2DM, and 16 long-standing patients with T2DM, Bhute et al. 139 found that opportunistic fungal pathogens such as Aspergillus and Candida were more abundant in newly diagnosed subjects compared with other groups. Jayasudha et al. 19 used metagenomic sequencing to characterize fungal structure in fecal samples from 21 individuals with T2DM and 30 healthy controls. They found that patients with T2DM had increased fungal richness and evenness in the gut compared to healthy controls. An increase in the abundance of known human pathogens (genus Candida, Kodamaea, and Meyerozyma) and a decrease in the phylum Mucoromycota were noted in the gut of T2DM subjects.
Apart from altered gut mycobiome, altered fungal abundance in oral cavity was also reported in the subjects with diabetes mellitus. C. albicans and C. glabrata were both detected in the oral cavity of patients with T1DM.140,141 Nowakowska et al. 141 suggested that C. glabrata resident in vagina and rectum was more than four times higher in women with diabetes than in nondiabetics. Significant associations were showed between glycemia and serum lipids with fungal abundance in patients with diabetes.137,141 Overall, these findings suggest that individuals with T2DM had gut mycobiome dysbiosis but whether this is the cause or consequence of the disease remains unknown. Most current studies are descriptive and longitudinal follow-up cohorts are important to delineate the importance of mycobiota dysbiosis in disease progression or complications in patients with diabetes mellitus.
Atherosclerosis is associated with metabolic diseases. 142 Gut mycobiota dysbiosis has also been shown to be related with the development of carotid atherosclerosis. 132 In a study of 33 subjects who had fecal samples collected for ITS amplicon sequencing, the subjects were divided into two groups of 12 nonobese subjects versus 21 obese subjects. The authors showed that the relative abundance of phylum Zygomycota and family Mucoraceae and Mucor racemosus in the fecal samples of participants were negatively correlated with the carotid intima-media thickness and to the risk of subclinical atherosclerosis (using the Framingham risk scores) in prospective follow-up. 132 A separate study compared gut mycobiome of 48 coronary atherosclerosis patients with healthy controls, and there was no significant difference in the fungal composition of fecal samples between both groups with ITS sequencing. However, the abundance of Thermoascus and species Malassezia restricta in the patients with coronary atherosclerosis was significantly lower than in healthy individuals, and the decrease of M. restricta might have a close association with lipid metabolism disorder in atherosclerosis patients. 143 Altogether these findings implied the emerging role of the gut mycobiota on atherosclerosis. As cardiovascular and metabolic risks vary in individuals and can also be influenced by external factors including diet and lifestyle, multiomics studies incorporating metabolomics, transcriptomics, and the overall gut microbiome are needed.
Therapeutic approaches targeting the gut mycobiome
Pathogenic fungi infections have been a major challenge to global health and leads to significant morbidity and mortality. Recently, study suggested that host adaptive immune system could suppress harmful fungal effectors of pathogenic fungi in the gut to improve their commensal fitness, so as to maintain intestinal homeostasis in healthy state. 144 However, pathogens such as C. albicans, C. neoformans, and Aspergillus fumigatus could present a threat on immunocompromised individuals. 145 In subjects with fungal infections, antifungal drugs are generally used as the first choice to clear the pathogens. Current antifungal drugs in clinical use consist of azoles (disruption of fungal ergosterol synthesis), polyenes (breakdown of membranes), echinocandins (inhibition of cell wall synthesis), and pyrimidines (inhibition of DNA synthesis and miscoding of RNA). 146 Invasive candidiasis is a frequent health-care-associated fungal infection caused by C. albicans, C. glabrata, and C. tropicalis. Mortality of this fungi infection is up to 40% annually.147,148 Amphotericin B deoxycholate and micafungin treatments are effective to control the infections.149,150 Recently alterations in the gut mycobiome have been reported in fecal samples of patients admitted to hospital with coronavirus disease (COVID-19) with increased proportions of opportunistic fungal pathogens, C. albicans, C. auris, and A. flavus compared with controls. Importantly, two respiratory-associated fungal pathogens, A. flavus and Aspergillus niger, were detected in fecal samples from a subset of patients with COVID-19, even after clearance of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) from nasopharyngeal samples and resolution of respiratory symptoms. 63 In sick individuals, Aspergillus sp. (A. fumigatus, A. penicillioides, A. niger and A. flavus) were detected in the endotracheal aspirate or bronchoalveolar lavage fluid imposing a high risk developing complicating invasive pulmonary aspergillosis. Antifungal therapies, such asvoriconazole and isavuconazole, were commonly used for preventing pulmonary aspergillosis and reducing mortality.151–153 However, toxicities and drug–drug interactions limit the utility of current antifungal drugs. Ubiquity use of antifungal drugs has contributed to our reliance on fungicides and the emergence of multidrug-resistant pathogenic fungi. 154
Diet could be one of the major driving force influencing gut fungal mycobiota structure. 90 A high-fat diet changed the gut fungal communities in murine models. It was reported that the abundances of the Alternaria, Saccharomyces, Septoriella, and Tilletiopsis genera were higher in mice administrated with normal chow compared with those fed with high-fat diet. 133 Moreover, diet rich in plant-derived carbohydrate can support Candida in the gut of Indian subjects, while saturated fatty acid and coconut oil–rich diet negatively correlated with the load of Candida in the gut.155–157 In addition, dietary short-chain fatty acids correlated with decreased colonization of Aspergillus spp. 90
Probiotics and prebiotics have been designed to provide health benefit. Multiple studies have identified several bacteria species with antifungal effects in vitro experiments, like Lactobacillus spp. and Bifidobacterium spp.158–160 In mice models, oral heat-killed Lactobacillus acidophilus (HKLA) and heat killed Lactobacillus casei could decrease the colonization of viable C. albicans in the tract. 161 Colitis would induce overgrowth of opportunistic yeast pathogen C. glabrata in murine gut. Mice with colitis treated with β-glucans presented a decreased level of C. glabrata in the gut compared the colitis mice. 162 Mycobiota regulation with probiotics and prebiotics in human study is rare, increasing evidences provide the possibility that the antagonistic relationships between bacterial and fungal species may decrease the perturbations and enhance the cross-talks in the gut to establish a balanced microbial community. 163
Other than bacteria, fungal probiotics could also confer beneficial effects to support human health. S. cerevisiae and S. boulardii are the most common yeast. S. boulardii has shown the potential abilities to alleviate gastrointestinal disorder caused by Helicobacter pylori, Salmonella, and Clostridium difficile.164–166 In the gut of patients with IBD, the environment may favor fungi over bacteria, leading to both fungal and bacterial microbiome dysbiosis.129,167 Guslandi has demonstrated that S. boulardii could be used to treat IBD effectively, modulating the microbial composition in the gut.168,169 However, whether S. boulardii was involved in the restoration of mycobiome composition in the gut remains unknown. Understanding how probiotics interplay with fungal community to maintain or restore a stable ecosystem and improve human health remains a challenge for future work.
Fecal microbiome transplantation (FMT) is the transfer of stool from a healthy individual to the gastrointestinal tract of another individual to re-establish the balance of microbiome. 170 Up to date, the effective application of FMT in treating patients with recurrent Clostridium difficile-associated diarrhea (CDI), 171 IBD, 172 irritable bowel syndrome (IBS) 173 have been successively demonstrated. The efficacy of FMT is also associated with mycobiota. CDI is accompanied by outgrowth of C. albicans and dysbiosis in fungal diversity. High levels of Saccharomyces and Aspergillus in donor stool were reported in FMT responders, while nonresponders were related to a high abundance of C. albicans. The results implied that FMI is a promising therapy to improve gut mycobiome dysbiosis in CDI patients, while the fungi also have a tight association with FMT treatment outcomes.
Future perspective
Here we review the existing literature on the human gut mycobiome in order to provide a comprehensive insight into both the methodologies available to research the gut mycobiota and also to highlight the latest research findings. We also draw on research into what is known about the human mycobiome composition at diseases and early life in order to provide both comparative insight and productive direction for future studies in this burgeoning research area. Studies performed so far have shown potential links of mycobiome in health and diseases, it is important to demonstrate the causation rather than association. An increasing body of evidence suggests the alteration of interkingdom microbial community alterations contributing to the detrimental consequences to the host. To expand our knowledge and obtain deeper insight into the role of the microbiome in health and disease, future studies should characterize the different microorganisms (bacteria, fungi, and viruses) in the same sample types and inter-kingdom microbial community. Future challenge will be to understand these interactions on both the molecular level and in their complexity. To meet this challenge, improved approaches and collaborations among bacteriologists, mycologists, immunologists, and clinicians are required to develop the foundation for personalized microbiome medicine. A better understanding of the fungi–bacteria–host interactions can allow identification of patients who are at risk and improvement of patient care by tailored manipulation of the microbiota.
Footnotes
Author contributions
Lin Zhang and Hui Zhan were joint first authors.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.