
Abstract
The ‘totality-of-the-evidence’ biosimilarity concept requires that sufficient structural, functional, nonclinical, and clinical data are acquired in a stepwise manner, to demonstrate that no clinically meaningful differences in quality, safety, or efficacy are observed compared with the reference product. We describe the totality of the evidence for PF-06438179/GP1111 (PF-SZ-IFX; IXIFI™ [infliximab-qbtx]/Zessly®) that supported its approval as an infliximab (IFX) biosimilar for all eligible indications of reference IFX (ref-IFX; Remicade®) in Europe and in the US. Analytical similarity involving in vitro assays capable of distinguishing structural or functional differences between PF-SZ-IFX and ref-IFX formed a foundation for the biosimilarity exercise. Differences identified in N-glycosylation and charge heterogeneity were found not to impact the results in in vitro biological assays reflective of the pharmacology underlying the mechanisms of action (tumor necrosis factor binding, reverse signaling, antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity) of IFX across disease indications. Similarity was assessed in a comparative clinical pharmacokinetic study and in a clinical efficacy and safety study in patients with rheumatoid arthritis, where therapeutic equivalence between PF-SZ-IFX and ref-IFX provided confirmatory evidence of biosimilarity, and, when coupled with the analytical similarity already established, supported extrapolation to all eligible disease indications of ref-IFX.
Introduction
Therapeutic antibodies currently occupy a central role in the treatment paradigm of many inflammatory diseases and cancers.1–3 Since it was first introduced in 1998, 4 the monoclonal antibody (mAb), infliximab ([IFX] marketed as Remicade®: Janssen Biotech, Inc., Horsham, PA, USA and Janssen Biologics B.V., Leiden, The Netherlands),5,6 has been widely used in the treatment of patients with immune-related diseases, including Crohn’s disease (CD) and ulcerative colitis (UC).7,8 Just as the expiry of patent protection, data and/or market exclusivities for a small-molecule drug often opens the way for manufacturers to provide generic versions, a similar potential opportunity arises at the end of respective protection and exclusivities for biologic drugs. Since biologics are created using highly specialized and proprietary processes in living cells, it is not possible to generate an identical copy of the originator biologic or reference product (RP), and so these new versions of the originator molecules are termed biosimilars or ‘similar biotherapeutic products’.9,10 The availability of biosimilars offers the potential for overall healthcare cost savings and increased patient access to treatments.11,12
The regulatory approval of biosimilars is governed by a distinct pathway,10,13,14 designed to establish that the quality, safety, and efficacy of the proposed biosimilar do not result in any clinically meaningful differences compared with its RP. This regulatory approach is reflected in the ‘totality-of-the-evidence’ concept, 13 whereby similarity to the RP with respect to a single property or area of testing (e.g., structural, functional, nonclinical, or clinical) is not sufficient by itself to establish biosimilarity. Only by evaluation of the entire data package is it possible to conclude that a biologic product can be approved as a biosimilar. As such, establishing biosimilarity takes a stepwise approach, with considerable reliance on the comparative structural and functional characterization of the proposed biosimilar and the originator RP, in addition to conducting a nonclinical and clinical assessment (Figure 1). 15 The success of this approach relies on the accumulation of knowledge and understanding of the proposed biosimilar and its RP, in order to interpret any differences identified between them, and to ensure that residual uncertainties arising at any step can be addressed during the development pathway.
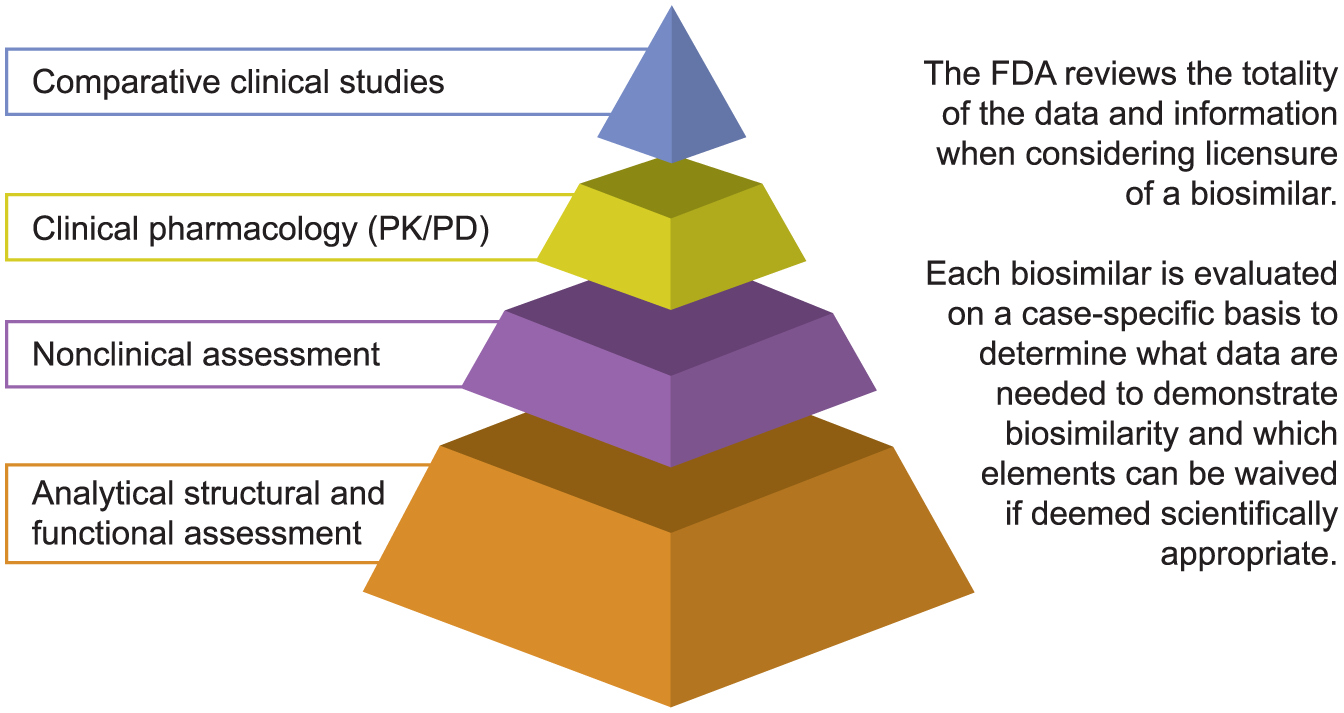
FDA ‘totality-of-the-evidence’ concept to demonstrating biosimilarity. 15
The European Medicines Agency (EMA) guidelines on biosimilars indicate that once biosimilarity in one indication has been demonstrated, provided there is appropriate scientific justification, extrapolation of the clinical data to other indications of the RP can be deemed acceptable. 14 In particular, consideration of extrapolation should be viewed on the basis of the totality of data obtained for the biosimilar (physicochemical and structural analyses, in vitro functional assessments, clinical efficacy, and safety and/or pharmacokinetic [PK]/pharmacodynamic [PD] data in one therapeutic indication), such that similar safety and efficacy of the biosimilar to the RP in the extrapolated indication can be expected. 16 Similarly, according to US Food and Drug Administration (FDA) guidance, 13 where there are data derived from a clinical study sufficient to demonstrate similarity in safety, purity, and potency in an appropriate condition of use, there is potential for a proposed biosimilar to be licensed for one or more additional conditions of use for which the RP is already authorized. To support extrapolation, there must be a robust scientific justification that addresses issues related to the indication in which the proposed biosimilar was assessed, and to the extrapolated indications. Scientific justification must include consideration of: the mechanism of action (MOA) in each indication for which authorization is sought; the PK and biodistribution of the proposed biosimilar across different patient populations (relevant measures of PD effect may yield valuable evidence to validate the MOA); differences in toxicity that can be anticipated in each indication and patient population; and any other factor that may affect the safety or efficacy of the proposed biosimilar in each condition and patient population for which licensure is sought.
PF-06438179/GP1111 ([PF-SZ-IFX] with market authorization as IXIFI™ [infliximab-qbtx]: Pfizer Inc, New York, NY, USA and as Zessly®: Sandoz GmbH, Kundl, Austria) is a biosimilar to Remicade® (ref-IFX) that was developed in line with the regulatory recommendations of the US FDA 13 and the EMA. 14 PF-SZ-IFX is approved in the US, 17 in the European Union, 18 and in Japan 19 for all indications held by ref-IFX and not covered by regulatory exclusivity. (The indication of pediatric UC for ref-IFX is currently protected by orphan drug exclusivity in the US.) This article describes the totality of the evidence demonstrating biosimilarity of PF-SZ-IFX with ref-IFX, highlighting the scientific rationale supporting extrapolation to all eligible indications authorized for ref-IFX, including inflammatory bowel disease.5,6
Structure and mechanism of action of IFX
IFX is a chimeric human–mouse type 1 immunoglobulin G (IgG1) kappa mAb. The crystallizable fragment (Fc) region of IFX consists of human IgG1, while the complementarity-determining region of the antigen-binding fragment (Fab) domain is derived from the mouse IgG1. 20
The importance of the cytokine tumor necrosis factor (TNF) in the pathophysiology of rheumatoid arthritis (RA), ankylosing spondylitis, psoriatic arthritis, plaque psoriasis, UC, and CD has been established by immunohistochemical evidence of increased expression of TNF in affected tissues in each disease state.21,22 The anti-inflammatory effects of IFX are mediated through multiple MOAs. 20 IFX binds to TNF (both soluble [sTNF] and transmembrane TNF [mTNF]) through the Fab region, and neutralizes the pro-inflammatory effects of TNF by blocking its interaction with TNF-type 1 (TNFR1 or p55) and TNFR2 (p75) cell surface receptors. 20 Binding of the Fab domain of IFX to sTNF results in disruption of TNF ligand–receptor signaling and inhibition of an inflammatory cascade, leading to downregulation of adhesion molecule expression, induction of apoptosis, activation, and secretion of other pro-inflammatory cytokines, and a reduction of the inflammatory infiltrate. 22 IFX binding to mTNF may also result in Fc domain-mediated mechanisms for neutralizing pro-inflammatory effects. 20
Binding and neutralization of TNF is common to anti-TNF mAbs, and this MOA is applicable across all disease indications of IFX (Table 1).8,20,23–38 However, binding of sTNF does not completely account for the effectiveness in IFX in the treatment of CD, and binding to mTNF appears to be of additional importance in this indication.23,24 The IFX/mTNF complex on the TNF-producing cell can block the binding to TNFR1/R2 on TNF-responsive cells, thereby inhibiting TNF-induced apoptosis. In a TNF-producing cell, binding of the Fab domain of IFX to mTNF can also result in ‘reverse signaling’ and a response such as cell apoptosis.
Mechanisms of action underlying control of TNF-mediated disease across indications of IFX.

ADCC, antibody-dependent cell-mediated cytotoxicity; AS, ankylosing spondylitis; CD, Crohn’s disease; CDC, complement-dependent cytotoxicity; Fab, fragment antigen-binding; Fc, crystallizable fragment; MOA, mechanism of action; mTNF, transmembrane TNF; PsA, psoriatic arthritis; PsO, plaque psoriasis; RA, rheumatoid arthritis; sTNF, soluble tumor necrosis factor; TNFR1, TNF-type 1 receptor; TNFR2, TNF-type 2 receptor; UC, ulcerative colitis.
Where IFX has bound to mTNF, it is also possible that a cytotoxic effect is produced via the Fc domain through either antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). The Fc domain of IFX binds to the Fcγ receptor (FcγR) on immune cells; FcγR comprises the subclasses I, II, and III, with further subtypes. For instance, IFX binds through the Fc domain to FcγRIIIa on effector (e.g., natural killer) cells. Upon activation and signaling, through ADCC, these cells lead to the lysis and apoptosis of target cells bound by the Fab domain. The binding affinity of the Fc region of IgG can vary with different polymorphisms of FcγIIIa. Two genetic variants of FcγRIIIa, 158V and 158F, are known to affect the binding to IgG and ADCC in vitro.39,40 FcγRIIIa polymorphisms have been reported not to be clinically significant to IFX treatment.41,42
While the prominence or the contributions of the MOAs of IFX outlined above may differ across disease indications, there is a consistent role for neutralization of the TNF-mediated inflammatory response via disruption of ligand-receptor interaction and function by IFX across a range of chronic, inflammatory disorders (Table 1).
Totality-of-the-evidence approach to establishing the biosimilarity of PF-SZ-IFX to ref-IFX
Based on the understanding of the MOA of IFX, and analysis of multiple batches of US-licensed ref-IFX (ref-IFX-US) and EU-approved ref-IFX (ref-IFX-EU), product quality attributes were identified that were considered to impact the PK, efficacy, and safety (including immunogenicity) of IFX. The totality-of-the-evidence approach was anchored on an in-depth assessment of the structural and functional attributes of PF-SZ-IFX, ref-IFX-US, and ref-IFX-EU, with respect to criteria set for establishing similarity. Comparison of ref-IFX-US and ref-IFX-EU provided a scientific bridge to subsequent clinical studies.
Structural and functional assessment
Multiple lots of ref-IFX-US and ref-IFX-EU purchased over several years were analyzed alongside PF-SZ-IFX drug substance and drug product in the similarity exercise, which is described in detail elsewhere 43 (also Conlon et al., in preparation). The results of the assessments (Figure 2) demonstrated a high degree of structural similarity between PF-SZ-IFX, ref-IFX-US, and ref-IFX-EU, with some minor differences observed that are discussed below.

Summary of analytical physicochemical and functional activity similarity assessment of PF-SZ-IFX and ref-IFX-US/ref-IFX-EU 43 (also Conlon et al., in preparation, 2019).
FcγRIIIa binding and subsequent ADCC can be influenced by the N-glycosylation pattern of the Fc region of IgG. 44 Results of N-linked glycan mapping by hydrophilic interaction liquid chromatography/mass spectrometry (MS) demonstrated that PF-SZ-IFX, ref-IFX-US, and ref-IFX-EU were highly similar with respect to the major level N-linked glycans (G0F and G1F) as well as the key N-linked glycan species: total afucosylated and terminal galactosylated. These key species are structural elements impacting ADCC and CDC, respectively. However, based on the results of ultra-high-resolution electrospray ionization quadrupole time-of-flight MS, used in the N-linked glycan mapping, sialylated glycans were found to be different between PF-SZ-IFX and ref-IFX (ref-IFX-EU and ref-IFX-US). N-acetylneuraminic acid (Neu5Ac) was observed as the predominant form in PF-SZ-IFX, while N-glycolylneuraminic acid (Neu5Gc) was observed as the predominant form in ref-IFX-EU and ref-IFX-US. Both N-linked glycan mapping and a sialic acid assay determined that the sialic acid was present at different levels of the total N-linked glycan structures for PF-SZ-IFX, and ref-IFX-EU/ref-IFX-US. In addition, trace levels of galactose-alpha-1,3-galactose (alpha-gal) extensions were also observed in ref-IFX-EU and ref-IFX-US that were not seen in PF-SZ-IFX. In vitro biological assays assessing FcγR binding, ADCC, and CDC did not show significant differences in activity for PF-SZ-IFX and ref-IFX-EU/ref-IFX-US. 43 Although differences in N-linked glycan species were observed between PF-SZ-IFX and ref-IFX-EU/ref-IFX-US, these differences were in line with expectations because of the different cell line used in the manufacture of PF-SZ-IFX (Chinese hamster ovary cells) and ref- IFX-EU/ref-IFX-US (mouse myeloma SP2/0 cells), and were not considered clinically relevant.45–47
Charge heterogeneity analysis determined that the level of basic species present differed significantly between PF-SZ-IFX and ref-IFX-EU/ref-IFX-US. 43 Extensive characterization, using MS techniques and enzymatic treatment, concluded this was due to the different level of heavy-chain C-terminal Lys present in each product. 43 C-terminal Lys is a commonly observed structural feature in the basic species of mAbs and is rapidly cleaved in vivo. 48 The different levels observed can be attributed to the cell lines used in the manufacture of the respective products. The structural differences in N-linked glycans and C-terminal Lys between products outlined above were not expected to impact efficacy or safety. 49
The extensive in vitro functional similarity assessment performed included biological assays reflecting the key attributes underlying the mechanisms of action of IFX across disease indications. In particular, PF-SZ-IFX displayed similar profiles to ref-IFX (ref-IFX-EU and ref-IFX-US) in binding to both sTNF and to mTNF.42,43 Similarity between products was also determined in terms of their Fc domain activity, since functional assays assessing ADCC and CDC showed comparable dose–response curves and relative potencies between products.42,43 These findings were ultimately substantiated by the similarity in efficacy and safety between PF-SZ-IFX and ref-IFX-EU in the comparative clinical study in patients with RA, 50 confirming that the structural differences described above were not clinically relevant and did not prevent a conclusion of biosimilarity of PF-SZ-IFX to ref-IFX.
Despite mAbs being highly complex, the analytical and in vitro biological assays used to characterize them are sufficiently sensitive to distinguish even minor variations between the originator RP and a proposed biosimilar. The need to establish whether any structural or functional differences observed as part of the analytical assessment have a clinical impact highlights the importance of the totality-of-the-evidence approach towards establishing biosimilarity.
Nonclinical assessment
Since assessment of the structural and functional characteristics (including in vitro pharmacology using assays that evaluated Fab-related biological activity and Fc-based functionality) demonstrated that PF-SZ-IFX was similar to ref-IFX-EU/ref-IFX-US, no in vivo efficacy studies were performed. Based on the similarity of PF-SZ-IFX to ref-IFX in structural and functional characteristics, the global regulatory guidance available at the time of assessment,13,16 and the lack of adverse toxicity in nonclinical toxicology studies with ref-IFX, 51 a single-dose toxicokinetic/tolerability study of intravenously administered PF-SZ-IFX or ref-IFX-EU to male rats was considered sufficient to address any residual concerns regarding the similarity of PF-SZ-IFX and ref-IFX. 43 A single dose of test article (PF-SZ-IFX or ref-IFX-EU) was considered adequate, based on the absence of toxicity observed in repeat-dose toxicity studies with ref-IFX. 51 This comparative study was conducted in rats because of ethical concerns associated with the use of chimpanzees (the only pharmacologically relevant species for toxicity testing with IFX), and the lack of toxicity observed in studies conducted with ref-IFX in rats, chimpanzees, mice, or rabbits. 51 In this single-dose toxicokinetics/tolerability study in rats, the effects of PF-SZ-IFX (10 mg/kg or 50 mg/kg) on mortality, clinical signs, body weight, anti-drug antibody (ADA) response, and exposure were similar to that of ref-IFX-EU. 43 These results demonstrated similarity of PF-SZ-IFX to ref-IFX-EU for nontarget mediated effects on tolerability, ADA response, and overall exposure. In addition to the findings from the structural and functional assessments, the results of this nonclinical toxicokinetics/tolerability study in male rats provided additional support for the totality of the evidence demonstrating biosimilarity of PF-SZ-IFX to ref-IFX.
Clinical similarity assessment
Clinical studies were performed to provide confirmatory evidence for the biosimilarity of PF-SZ-IFX and ref-IFX-US/ref-IFX-EU established from the structural and functional assessments and based on the results of the nonclinical in vivo study. The clinical development program that confirmed the similarity of PF-SZ-IFX to ref-IFX comprised a phase I PK similarity study in healthy subjects, 52 and a comparative efficacy and safety study in patients with moderately to severely active RA (Figure 3). 50

Pharmacokinetics
In PK studies conducted in various patient populations, it was previously shown that following single or repeated intravenous administration of ref-IFX at doses of 3–20 mg/kg, there was a linear relationship between the dose administered and the maximum serum concentration (Cmax) and area under concentration–time curve (AUC) values.5,53 The median terminal half-life ranged from 7.7 to 9.5 days, after single doses at 3–10 mg/kg in patients with RA, 5 mg/kg in patients with CD, and 3–5 mg/kg in patients with plaque psoriasis. The formation of ADAs is known to affect the PK of IFX in patients with RA. 54 Higher serum IFX concentrations at a low IFX dose (1 mg/kg) in patients with RA receiving repeated IFX dosing with concomitant administration of methotrexate (MTX) can be ascribed to suppression of ADA formation, and the IFX concentration being unaffected. 55 While the PK of IFX is similar across the approved indications, the extent of ADA formation, the use of—and response to—concomitant immunosuppressants, and their impact on PK parameters may differ.31,32,55–58 Nevertheless, it was anticipated that the PK profile for PF-SZ-IFX and ref-IFX should be similar, regardless of the patient setting, because of the similarity in structural and functional in vitro data that had already been established.
A phase I, double-blind, parallel-group, three-arm trial (Study B5371001) was conducted to demonstrate PK similarity of PF-SZ-IFX to ref-IFX-EU and to ref-IFX-US, and of ref-IFX-EU to ref-IFX-US. 52 Healthy adult subjects, randomized to PF-SZ-IFX, ref-IFX-EU, or ref-IFX-US, received a single 10 mg/kg treatment dose by intravenous infusion. The primary objective was to assess PK similarity based on the PK parameters, Cmax, AUC from time 0 to the last time point measurable concentration (AUCT), and AUC from time 0 to infinity (AUCinf). PK assessments were performed over 8 weeks, and safety and immunogenicity evaluations were determined over 12 weeks. PK similarity of PF-SZ-IFX to both ref-IFX-US and ref-IFX-EU, and of ref- IFX-EU to ref-IFX-US, was demonstrated in this study. Results showed that for the PK parameters, Cmax, AUCT, AUCinf, the 90% confidence interval (CI) of point estimates of the test-to-reference ratios were contained in the prespecified equivalence range of 80–125% (Figure 4).
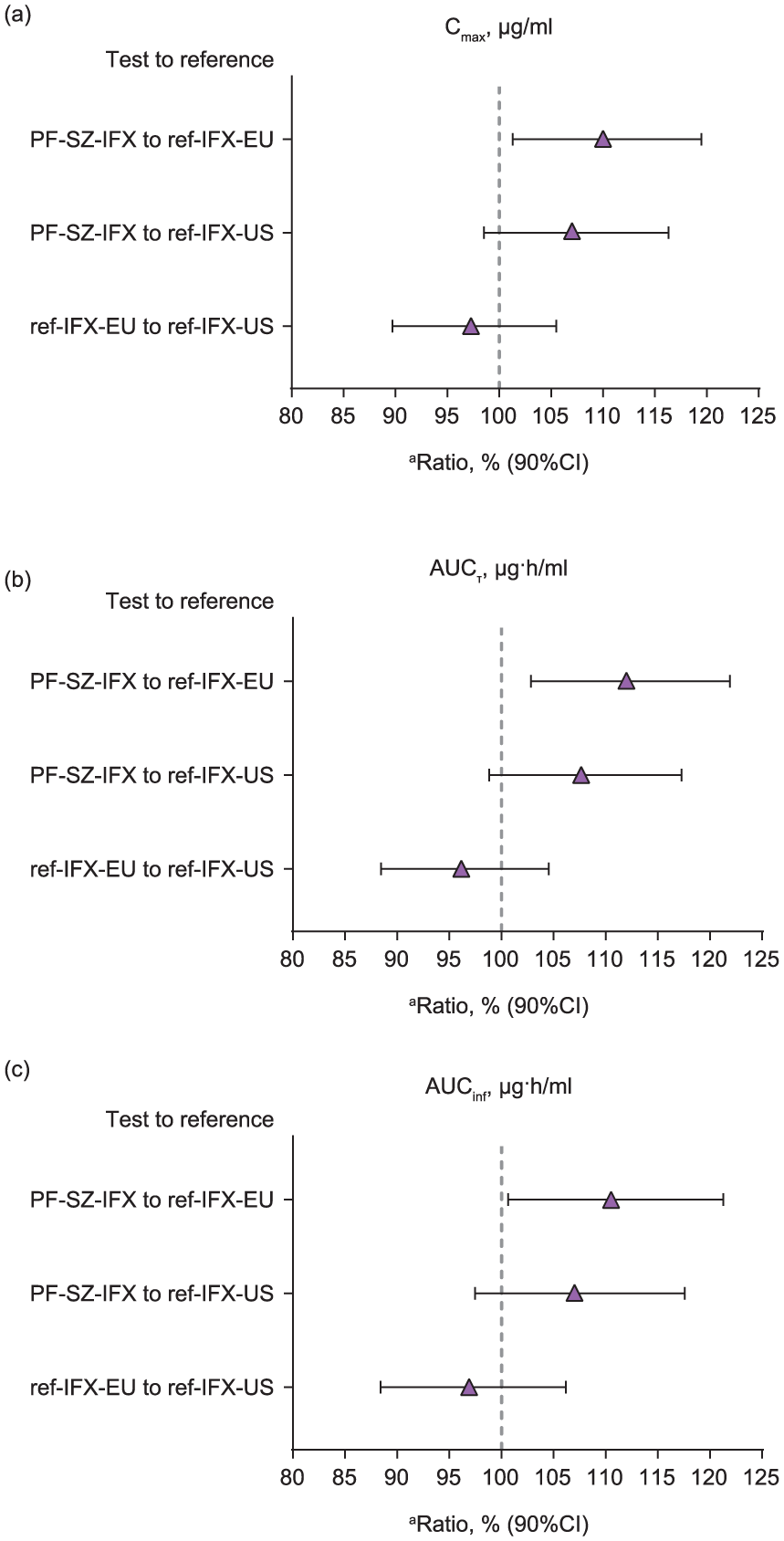
Statistical comparison between test and reference products of PK exposure parameters: (a) Cmax; (b) AUCT; (c) AUCinf.. 52
The PK of PF-SZ-IFX and ref-IFX-EU were also assessed as part of the comparative efficacy and safety trial in patients with RA (Study B5371002). 50 Serum PF-SZ-IFX and ref-IFX-EU concentrations were similar, and the impact of ADA on PK in ADA-positive patients was similar between treatment arms (Figure 5). As anticipated, the serum concentrations of PF-SZ-IFX and ref-IFX-EU were lower in ADA-positive patients compared with ADA-negative patients. The findings from a population PK (PopPK) analysis of Study B5371002 (Palaparthy et al., manuscript submitted for publication, 2019) were consistent with those of previously reported PopPK analyses for ref-IFX. The PK of both ref-IFX-EU and PF-SZ-IFX were adequately described by a two-compartmental model with linear elimination from the central compartment, with no appreciable differences between the PK parameters of PF-SZ-IFX and ref-IFX-EU in this patient population. This analysis determined that the covariates that significantly influenced the PK parameter variability of ref-IFX-EU and PF-SZ-IFX were the same—namely baseline body weight, sex, and ADA titers—and provided further supporting evidence of PK similarity between the two products (Palaparthy et al., manuscript submitted for publication, 2019).
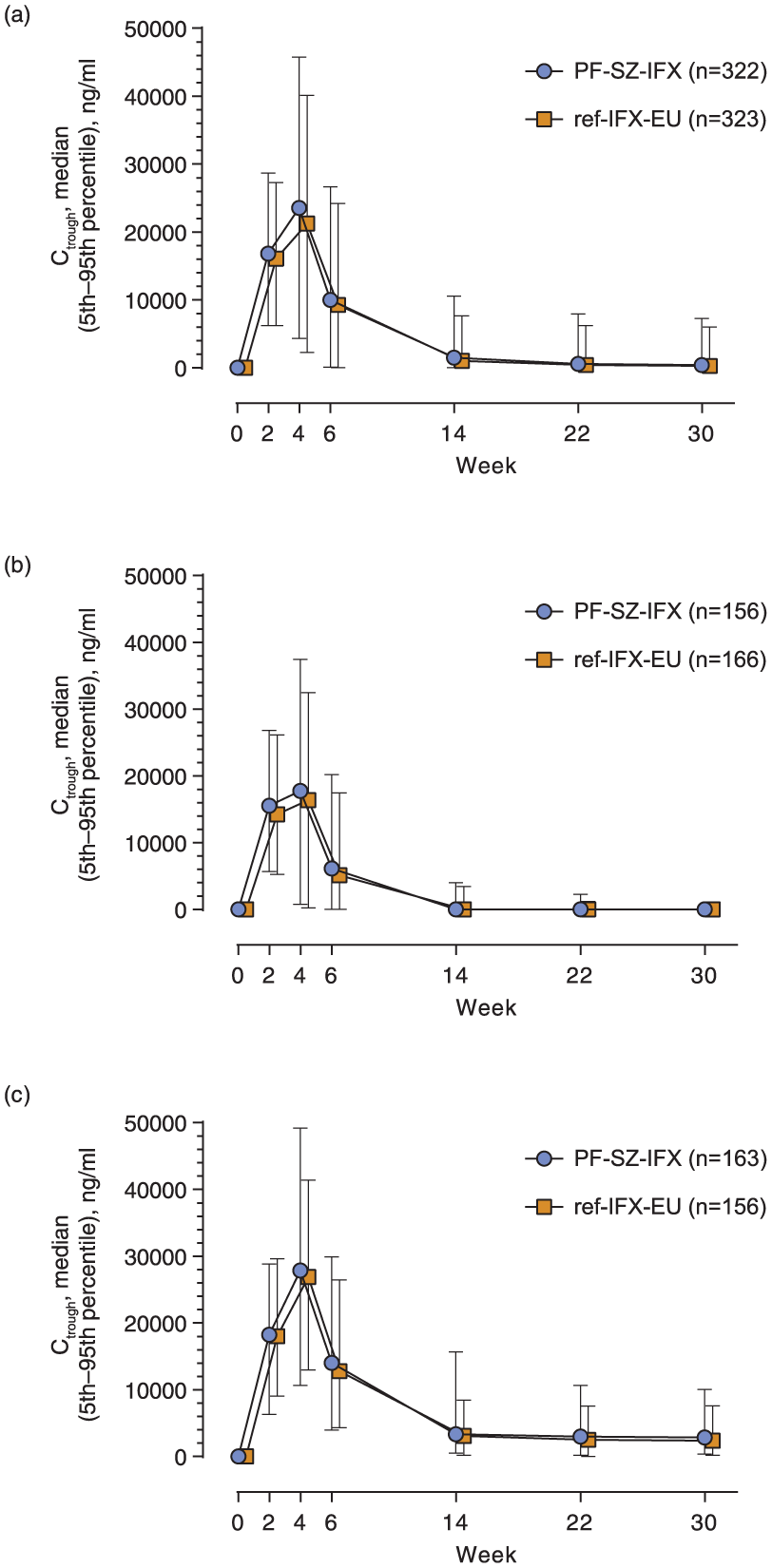
Serum PF-SZ-IFX and ref-IFX-EU Ctrough concentrations by study visit and ADA status (PK population): (a) All patients; (b) ADA-positive patients; (c) ADA-negative patients. 50
Clinical efficacy
An RA patient population was selected for the comparative efficacy and safety trial (Study B5371002) since it provides a sensitive setting to detect differences among effective treatments, and it has a large clinical utilization, immunogenicity, and safety experience amongst the various IFX-licensed indications. 59 The efficacy and safety of PF-SZ-IFX and ref-IFX-EU were compared in RA patients with an inadequate response to MTX. 50 In both the intent-to-treat and per-protocol populations, the two-sided 95% CIs and 90% CIs of the treatment difference in the 20% improvement in American College of Rheumatology (ACR) criteria (ACR20) response rate at week 14 were entirely contained within the symmetric equivalence margin (−13.5 to 13.5%) and the asymmetric equivalence margin (−12.0 to 15.0%), respectively, demonstrating therapeutic equivalence (similarity) between PF-SZ-IFX and ref-IFX-EU. 50 Similar responses between PF-SZ-IFX and ref-IFX-EU were also observed at each study visit up to week 30, as measured by: ACR20/50/70 (Figure 6a); Disease Activity Score (DAS)-28, four components based on high-sensitivity C-reactive protein (Figure 6b); European League Against Rheumatism (EULAR) response; DAS remission; ACR/EULAR remission; and individual ACR parameters, including the health assessment questionnaire-disability index. 50

ACR20, ACR50, and ACR70 response rates (a), and mean (± SE) change from baseline in DAS28-CRP (b) for PF-SZ-IFX and ref-IFX-EU by visit (ITT population). 50
Clinical safety
The safety population in Study B5371001 comprised 146 subjects: 49 subjects randomized to the PF-SZ-IFX arm, 48 subjects randomized to the ref-IFX-US arm, and 49 subjects randomized to the ref-IFX-EU arm. 52 All treatments were found to be generally safe and well tolerated.
The safety profiles of PF-SZ-IFX and ref-IFX-EU in Study B5371002 were found to be similar during the initial 30-week treatment period. A total of 185 (57.3%) and 176 (54.0%) patients reported all-causality treatment-emergent adverse events (TEAEs) in the PF-SZ-IFX and ref-IFX-EU treatment arms, respectively (Figure 7). ‘Infections and infestations’ was the system organ class (SOC) with the highest number of patients who had TEAEs (86 [26.6%] patients receiving PF-SZ-IFX, and 72 [22.1%] patients receiving ref-IFX-EU). The most frequently reported TEAE was infusion-related reaction (IRR; 19 [5.9%] patients receiving PF-SZ-IFX, and 21 [6.4%] patients receiving ref- IFX-EU). In the PF-SZ-IFX and ref-IFX-EU arms, TEAEs reported as potentially related to study treatment by the investigator (treatment-related TEAEs) occurred in 81 (25.1%) and 75 (23.0%) patients, respectively. ‘Infections and infestations’ was the SOC with the highest percentage of patients who experienced treatment-related TEAEs (occurring in 28 [8.7%] and 22 [6.7%] patients in the PF-SZ-IFX and ref-IFX-EU arms, respectively), with IRR being the most frequently reported treatment-related TEAE (17 [5.3%] and 20 [6.1%] patients, respectively). Similar proportions of patients in the PF-SZ-IFX and ref-IFX-EU treatment arms (16 [5.0%] and 20 [6.1%] patients, respectively) reported serious adverse events. Upon review of all safety data from the initial 30-week treatment period, the safety profile of PF-SZ-IFX was found to be similar to that of ref-IFX-EU.

All-causality TEAEs (safety population). 50
Clinical immunogenicity
The occurrence of immunogenicity to biologics is complex and influenced by various intrinsic and extrinsic factors. 60 The factors influencing the development of ADAs can be related to treatment (e.g., dose and duration, frequency, route of administration); product (e.g., amino acid sequence or glycosylation pattern); process (e.g., manufacturing, storage, handling, impurity profile); and patient population (e.g., genetic predisposition, immunosuppressed). Incidence of ADAs can also vary between studies due to the format, sensitivity, and specificity of the assay, as well as aspects such as the threshold used or the sampling schedule. 61 While it is known that the immunogenicity of ref-IFX is not the same across all patient populations within its licensed indications, 6 it was anticipated that the immunogenicity profiles for PF-SZ-IFX and ref-IFX should be similar, irrespective of the patient setting, since the intrinsic and extrinsic factors would be the same.
All three treatment groups (PF-SZ-IFX, ref-IFX-EU, and ref-IFX-US) in Study B5371001 had an overall comparable incidence of ADA. 52 In Study B5371002, conducted in patients with RA, the overall proportions of ADA-positive patients to week 30 for the PF-SZ-IFX (48.6%) and ref-IFX-EU (51.2%) arms were similar. 50 Approximately 80% of all ADA-positive patients overall also tested positive for neutralizing antibody (NAb); the ADA/NAb results were balanced between treatment arms. 50 Overall, immunogenicity assessments were consistent with the findings from the analytical structural and functional, and nonclinical, assessments in that there were no meaningful differences between PF-SZ-IFX and ref-IFX-EU or ref-IFX-US. Moreover, these findings confirmed that the immunogenicity profile of PF-SZ-IFX was unaffected by being manufactured in a different cell line to that used to produce ref-IFX.
Extrapolation of indications
The biosimilarity demonstrated between PF-SZ-IFX and ref-IFX, and the established evidence of an MOA for IFX consistent with ligand–receptor interaction and function across all the licensed indications of ref-IFX, provides scientific justification for the use of PF-SZ-IFX in all clinical indications of ref-IFX, including those not specifically studied in the PF-SZ-IFX clinical program. Additionally, data established with ref-IFX in various subpopulations (such as those based on age, gender, ethnicity, comorbidities, concurrent therapies), as well as data from its use at different dosages and in combination regimens, are also extrapolated for PF-SZ-IFX. Since the scientific justification for extrapolation is based on the assertion that PF-SZ-IFX has been shown to be similar to ref-IFX through multiple lines of evidence (including a clinical trial in a single indication), PF-SZ-IFX is expected to have similar clinical activity to ref-IFX in all clinical adult and pediatric settings in which ref-IFX has been evaluated.
Conclusion
The totality-of-the-evidence approach for PF-SZ-IFX was built on regulatory and scientific principles. Extensive studies to determine the structural and functional characteristics of PF-SZ-IFX provided the foundation for the similarity assessment.42,43 Differences between PF-SZ-IFX, ref-IFX-US, and ref-IFX-EU were characterized and determined not to impact the in vitro biological activity of the three products or to be clinically relevant. The totality of the evidence obtained—comprising structural, functional, nonclinical, and clinical data—enabled a determination of biosimilarity between PF-SZ-IFX and ref-IFX, and was consistent with the regulatory requirements13,14 for the approval of PF-SZ-IFX across all the eligible indications of ref-IFX. The biosimilarity established for PF-SZ-IFX to ref-IFX justifies extrapolation of the established benefit–risk of ref-IFX to PF-SZ-IFX, and supports the expectation that they will behave as one another in the clinical setting across all indications.
Footnotes
Acknowledgements
Medical writing support was provided by Iain McDonald PhD, Engage Scientific Solutions, and was funded by Pfizer Inc.
Funding
Medical writing support for this review article was funded by Pfizer Inc.
Conflict of interest statement
Joseph E. McClellan, Hugh D. Conlon, Michael W. Bolt, Vatche Kalfayan, Rameshraja Palaparthy, Muhammad I. Rehman, and Carol F. Kirchhoff are employees of and hold stock in Pfizer Inc.