
Abstract
The application of next-generation sequencing in clinical practice is increasing as accuracy and interpretation have improved and the cost continues to decline rapidly. Cell-free DNA is a unique source for next-generation sequencing that could change routine clinical practice in gastroenterology and hepatology. Testing of cell-free DNA in blood and fecal samples is an easy, rapid, and noninvasive method to assess for premalignant, malignant, metabolic, infectious, inflammatory, and autoimmune gastrointestinal and liver diseases. In this review, we describe cell-free DNA technologies, current applications of cell-free DNA testing, and proposed cell-free DNA targets for gastrointestinal and hepatic diseases, with a specific focus on malignancy. In addition, we provide commentary on how cell-free DNA can be integrated into clinical practice and help guide diagnosis, prognosis, disease management, and therapeutic response.
Introduction
The existence of extracellular DNA circulating within the blood, termed cell-free DNA (cfDNA), is a promising source of diagnostic material for the management of disease including digestive disorders. Despite the discovery of cfDNA over 60 years ago, it has only recently become feasible to use this type of DNA in the clinical setting. Currently, the main clinical utility of cfDNA testing is for prenatal testing of fetal aneuploidy and sex determination, to gage organ transplant rejection, as well as in oncology for diagnostic and therapeutic purposes as a sensitive and specific surrogate to tissue biopsy. Although this nascent technology has great promise to act as a ‘liquid biopsy’ and noninvasively diagnose disease, there remains many challenges in finding and validating clinically meaningful cfDNA biomarkers.
In this article, we review current advances in and applications of cfDNA technologies, describe how these methodologies can aid in the diagnosis and prognosis of digestive diseases, focusing on malignancy, and discuss the limitations and challenges of these applications.
Background
By definition, cfDNA is extracellular and is found in the blood as well as in other bodily fluids such as urine, bile, pancreatic secretions, or peritoneal fluid (Figure 1). However, the term ‘cell-free’ should not be interpreted that the DNA is circulating completely freely as cfDNA can be found adherent to antibodies, red blood cell membranes, proteins, particularly nucleosomes, in single-stranded, double-stranded, and triple-stranded DNA forms, in complex with RNA, as well as in adducts to proteins. cfDNA should also be differentiated from circulating cells such as circulating tumor cells, which are a source of diagnostic DNA but are not cell-free and therefore must be handled differently for laboratory purposes. In addition, cfDNA should be distinguished from circulating RNA such as messenger or microRNA can also be found in the blood and be used to diagnose diseases and this topic is reviewed elsewhere. 1
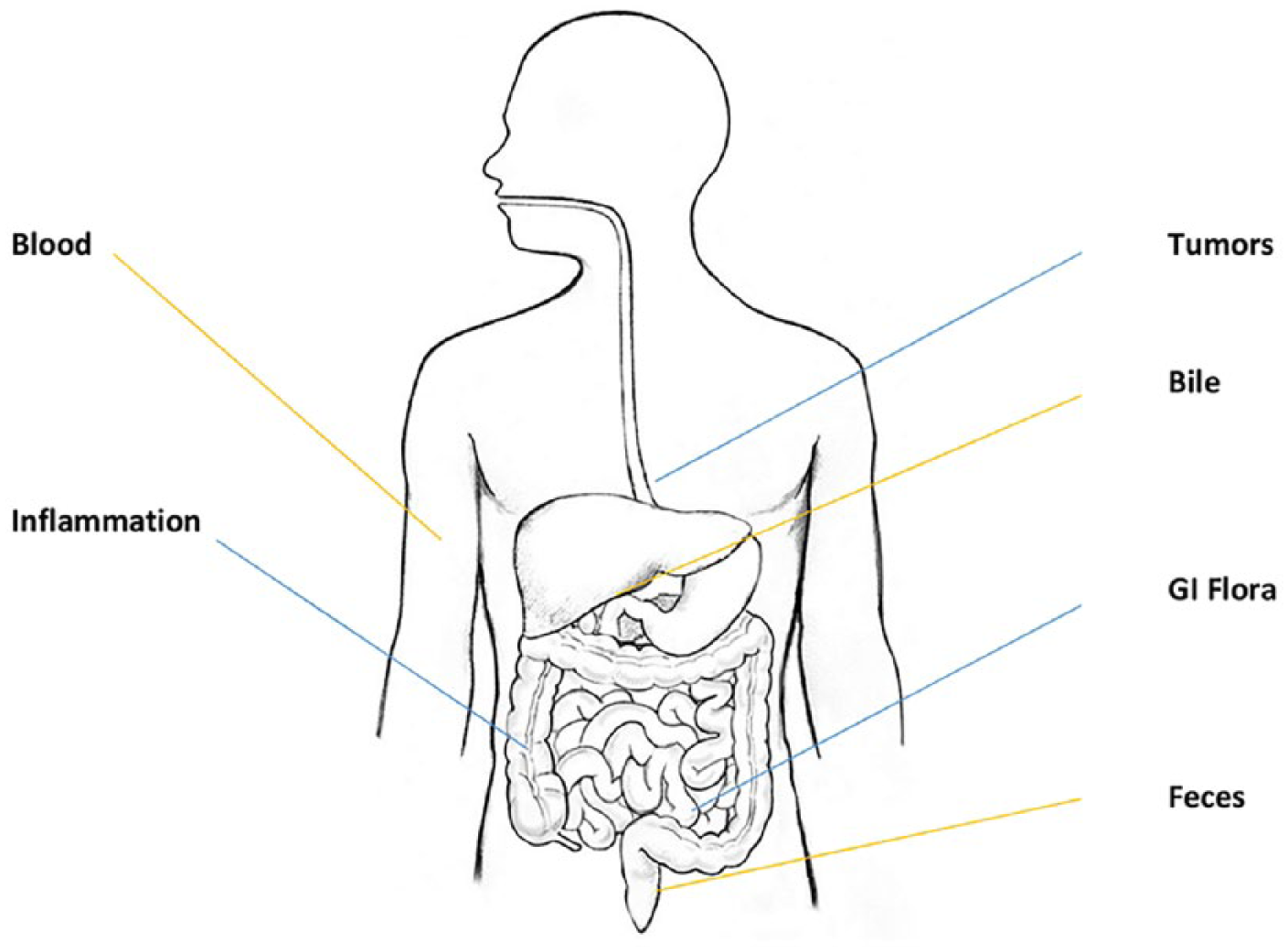
Sources of cell-free DNA (cfDNA).
There are several sources of cfDNA, and it can be found in the plasma of healthy persons, but an increased concentration of cfDNA can be found in several disease states, especially in inflammatory conditions such as systemic lupus erythematosus as well as with many cancers. Moreover, viral and bacterial DNA can be found circulating in the blood during both healthy and pathologic conditions. In fact, gastrointestinal-derived bacterial DNA can be found circulating in the blood of healthy subjects as well as in increased concentrations in the blood of subjects with inflammatory bowel disease or liver cirrhosis.2,3 The majority of cfDNA research has been completed on blood cfDNA, most commonly in plasma, rather than serum. Far fewer studies have been performed on other bodily fluids such as bile, pancreatic juices, urine, or stool, and therefore this review focuses on circulating cfDNA in the blood.
Despite the discovery of cfDNA in the many varied forms described above, there is still controversy regarding some of its basic properties such as the size ranges found circulating in the blood as well as the mechanism of release of DNA into the extracellular space. Undoubtedly, apoptosis and necrosis of cells are a source, however, there is speculation that even healthy cells may release genomic DNA. Size determination has been hindered by the fragile nature of free, unprotected, DNA and by artifacts that can be introduced by differences in handling and processing of samples and by laboratory equipment. Methods as varied as simple gel electrophoresis, Sanger sequencing, and electron and atomic force microscopy have produced wildly variable estimates in the average cfDNA size. The lower and upper limits of size ranges from a dozen base pairs to tens of thousands of kilobases.
Regardless of size, it cannot be disputed that the total levels cfDNA can be accurately and reliably measured and are found to vary between healthy and certain disease states. Moreover, with recent technological advances the full breadth of the variations and aberrations seen within nuclear genomic DNA can also be reliably measured in cfDNA, including single or multiple base pair substitutions, insertions, deletions, copy number variations and methylation. 4
Current advances and applications in cfDNA technologies
In the past, there were two main challenges preventing robust analysis of cfDNA, namely, low concentration and low allele frequencies. Indeed, the overall concentration of total cfDNA is low within the blood and the aberrant allele being tested for might only constitute a small fraction of the total cfDNA. In these scenarios, the mutant allele is obscured during traditional Sanger sequencing by the overwhelming copies of the normal allele. For instance, plasma concentrations of total cfDNA in a cancer patient may be on the order of 2 µg/ml and the frequency of a mutant allele such as KRAS 35G>A (KRasG12D) may only constitute 0.01% of all the circulating DNA (the range of plasma cfDNA in cancer patients in many studies falls ~20 ng/ml–2 µg/ml, as compared with ~0.1–20 ng/ml of cfDNA in the plasma of healthy controls in most studies).2,5
Currently, the challenges of low concentrations and low allele frequencies of cfDNA have been solved by new technological advances. These methods are outlined in Figure 2. Beyond traditional polymerase chain reaction (PCR), the first group of methods involve a priori knowledge of the specific mutant allele being tested. One such method is amplification-refractory mutation system (ARMS) PCR, also known as allele-specific PCR (ASPCR), which has been shown to reliably detect fewer than 100 copies of per milliliter of plasma (subnanogram range).5,6 Assays based on the same principle as ARMS have also been developed using fluorescent probes and quenchers to allow for real-time quantitation.

Methods of detecting cell-free DNA. ARMS, amplification-refractory mutation system; NGS, next generation sequencing; PCR, polymerase chain reaction; QPCR, quantitative real-time PCR.
Another method for assaying known mutations is to block the amplification of wildtype allele with ‘clamps’ that hybridize to the wildtype allele and block DNA polymerase, or by destruction of the wildtype allele with restriction enzymes. Many ingenious clamps have been designed to efficiently and stably hybridize to target sequences without themselves acting as primers, including protein nucleic acids, 3’-end locked nucleic acids, and xenonucleic acids. These methods allow for the detection of mutant alleles starting with just 15 femtogram (millionth of a nanogram) of genomic DNA dissolved 1:1000 with wildtype DNA. 7
Finally, a method that both detects low-frequency alleles and is also able to quantify the exact frequency of the mutant/variant allele is digital droplet PCR. This method relies on separating each strand of cfDNA into its own reaction vesicle or droplet. This is usually accomplished through emulsification of the sample DNA and PCR reagents within a single PCR tube. With only one strand of DNA per reaction there is no chance of competition between wildtype and mutant sequences and, thus, each droplet results in a binary reading of 1 for a positive result or a 0 for a negative result and hence the ‘digital’ reading. All of the droplets are read, usually through a fluorescent PCR reaction, and the results tallied so that the exact frequency of a mutant allele can be calculated. 8 One such method is BEAMing, which stems from an abbreviation of the components of the process (beads, emulsion, amplification, and magnetics). 9
The main advantage of all the aforementioned assays is that they are highly sensitive, however, they rely on knowledge of the specific mutation and, thus, must be multiplexed to assay for other mutations. The second group of cfDNA assays do not involve a priori knowledge of the specific mutation or variant. These assays rely on massively parallel sequencing or ‘next-generation sequencing’ (NGS) in which all the collected cfDNA is sequenced. These assays all rely on fragmentation of DNA into smaller pieces, separation of individual strands into single reaction vessels, and some form of readout of the sequence, typically optical readout of fluorescently labeled nucleotides added during a polymerase reaction. However, currently, most NGS systems have multiple limitations with regards to analyzing cfDNA. First, because these systems rely on multiple short reads to assemble larger sequences, they do not have the sensitivity to detect small (<1%) copy numbers of aberrant DNA. Second, massive amounts of data are generated and may result in the detection of multiple mutations of unclear significance for clinicians, and distress for patients. Third, these platforms tend to be much more costly than targeted tests. In addition to sequence variation, copy number can also be assayed for in cfDNA. Methods for this detection generally rely on quantitative real-time PCRs (QPCRs) or NGS. Clinically, copy number variants are used to assay for oncogene amplifications such as NMYC or HER2 amplifications, or to assess for duplications and deletions of whole portions of chromosomes.
Similarly, methylation status can be assayed for with any of the previously described methods, the main difference being that samples are bisulfite treated beforehand, which distinguishes methylated cytosine from nonmethylated cytosine. A hidden power of this method is that different tissues have characteristic methylation signatures and, therefore, cfDNA from different tissues may be distinguished in the peripheral blood, allowing for specific analysis of cfDNA originating from the gastrointestinal tissue of interest. More commonly, methylation status of given genes can be used to differentiate healthy versus disease states, which we describe in the next section. Though this technique can be powerful, one major limitation of bisulfite treatment is the loss of significant fragments of cfDNA, particularly smaller (<150 bp) portions that make up a large portion of the cfDNA. Current bisulfite techniques result in recovery of anywhere between 13% and 69% of the input DNA depending on the commercial kit used. 10
Specific uses of cfDNA testing in gastrointestinal and liver diseases
A laboratory technique is only useful if there are specific clinical applications; and in this section we will review possible cfDNA targets based on research and current clinical practice, with a summary of putative targets in gastroenterology and hepatology listed in Table 1.
Summary of putative targets in gastroenterology and hepatology.
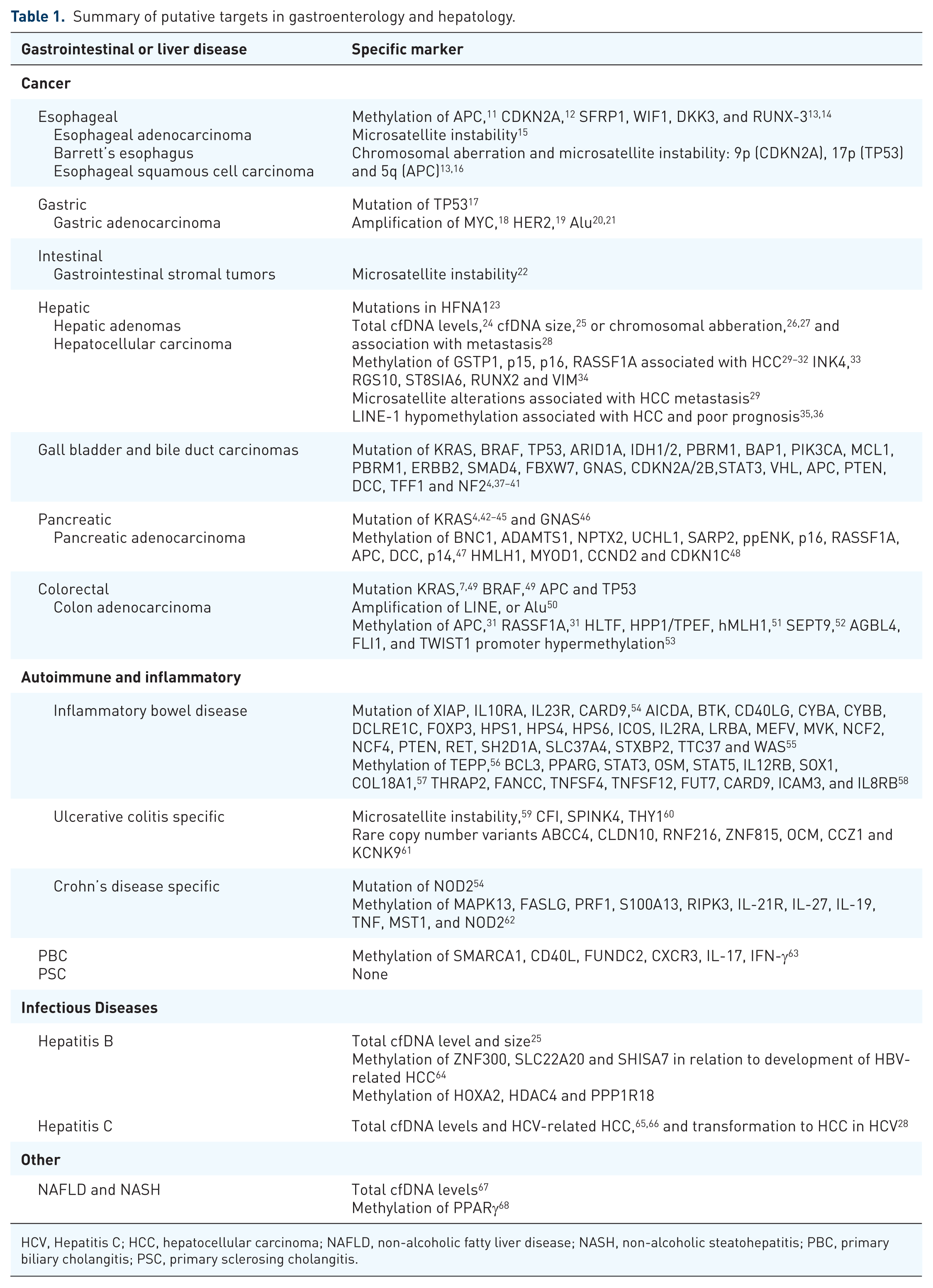
HCV, Hepatitis C; HCC, hepatocellular carcinoma; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PBC, primary biliary cholangitis; PSC, primary sclerosing cholangitis.
By far, the area with the most potential targets is cancer, and the rest of this review will focus on malignancy, though included in Table 1 are putative cfDNA markers in other digestive diseases. Not only is there a plethora of research into cancer genetics, but also the mutational process that drives cancer makes cfDNA approaches a low-hanging fruit. As mentioned previously, many studies have indicated that the total amount of cfDNA circulating in the blood is increased in many cancers and inflammatory conditions.6,24 Thus, testing for the concentration of cfDNA could be thought of as a general marker akin to the erythrocyte sedimentation rate as a surrogate for inflammation.
Upper gastrointestinal cancers
A use of cfDNA could be to differentiate malignant from premalignant conditions. For instance, patients with Barrett’s esophagus and esophageal adenocarcinoma could be differentiated by a 911 gene signature from a plasma cfDNA methylome proof of concept study. 69 Theoretically, this type of signature could be valuable to risk stratify patients with severe gastroesophageal reflux disease (GERD) or Barrett’s esophagus into high and low risk groups for endoscopic screening. Similar targets exist in gastric adenocarcinomas and gastrointestinal stromal tumor (GIST), which could be used to screen for cancer in at risk individuals with concerning symptoms, or to differentiate benign from malignant gastric ulcers (Table 1).
Hepatobiliary and pancreatic cancers
A clinical application of cfDNA could be to diagnose and risk-stratify patients with hepatic masses. For instance, it can be challenging to differentiate atypical focal nodular hyperplasia, hepatocellular adenoma, and hepatocellular carcinoma (HCC) when radiographically ambiguous. Moreover, patients with hepatocellular adenoma need to be screened for malignant transformation to HCC. There are a number of putative markers that can diagnose and differentiate these tumors, some of which have already been found in cfDNA (Table 1). These markers can be used to risk-stratify patients to identify those who require biopsy, ablation, or surgical resection. HFNA1 mutations are found in 30–40% of hepatocellular adenomas and can be used to diagnose these lesions from other tumors when imaging techniques are equivocal. 23 In addition, mutation of β-catenin in hepatocellular adenoma is strongly associated with malignant transformation into HCC, therefore identification of the minority of hepatocellular adenomas with β-catenin mutation via cfDNA could help risk-stratify patients and prioritize surgical resection or ablation of those patients with the relevant mutation.23,70 This also applies to HCC screening in patients with hepatitis C (HCV) or cirrhosis, and has even been used to diagnose HCC in patients without elevated levels of traditional markers such as alpha fetoprotein. 34
Similarly, radiographic identification of gallbladder cancer can be challenging and may mimic cholecystitis, adenomyomatosis, or polyps of the gallbladder. Pilot studies have demonstrated that the quantity of cfDNA is significantly different between inflammatory conditions and gallbladder carcinoma, suggesting use as a biomarker. 71 Biliary strictures or pancreatic cysts that are equivocal for malignancy can be evaluated with endoscopic retrograde cholangiopancreatography (ERCP) brush samples or aspirated pancreatic cyst fluid during endoscopic ultrasound. In either case, cytology is valuable and specific for detecting malignancy but not sensitive. cfDNA from biliary brush samples or cyst aspirates has been shown to increase the sensitivity of detecting malignancy without decreasing specificity when detecting oncogenes such as mutant KRAS or loss of various tumor-suppressors.37,38,72
Lower gastrointestinal cancers
In colorectal cancer, cfDNA has been used to distinguish patients with benign polyps from those with colorectal cancer, as well as to predict progression and treatment resistance. 50 For instance, the total level of cfDNA correlates with overall and progression-free survival as closely as the patient’s performance status, and surprisingly even more so than the number of metastatic sites.6,49,73 Moreover, cfDNA can be used to identify resistance to targeted therapies prior radiographic findings of progression. For instance, patients that were previously wildtype for KRAS mutation and treated with the epidermal growth factor receptor (EGFR) inhibitors, were identified to have KRAS mutation in cfDNA and, therefore, resistance to EGFR inhibition, at times months prior to radiographic progression.74–76
Integrating cfDNA testing into current gastrointestinal and liver diseases diagnostics
In the United States, current preventative screening tests for gastrointestinal and liver diseases are limited to interval colonoscopy for colon cancers and one-time HCV screening for individuals born between 1945 and 1965. However, less than two-thirds of eligible Americans are screened for colon cancer per guidelines, with likely one of the most common reasons being unwillingness to undergo colonoscopy. Less-invasive screening techniques such as multitarget stool DNA testing (i.e. Cologuard®) are effective and work by testing for aberrantly methylated BMP3 and NDR54 promoter regions, mutant KRAS, and quantitative immunochemical assay for human hemoglobin. 77 Other similar colorectal cancer screening panels have also been devised with targets such as SFRP2 methylation, mutation in APC, TP53, KRAS and microsatellite instability markers with relatively high sensitivity and specificity.78–80 The DNA used in these tests likely represents a combination of cellular DNA from sloughed cells as well as cfDNA, thus assaying the entire gastrointestinal tract at once.
The approach used for noninvasive colon cancer screening, with risk-stratification of patients into those who require colonoscopy versus those who can continue with interval noninvasive screening, could be extended to other gastrointestinal and liver diseases, and specifically applied to diseases diagnosed or treated with esophagogastroduodenoscopy (EGD) or ERCP. For instance, this approach can be used to diagnose celiac disease, for which ~3–5% of patients do not have anti-transglutaminase or anti-endomysial IgA antibodies. Though there is no published cfDNA signature for celiac disease, it is conceivable that a plasma cfDNA signature exists to reflect the T-cell and B-cell activity, which drives small bowel inflammation in celiac disease. A positive cfDNA test would then be followed by a confirmatory EGD.
Another example is pancreatic cancer for which there is currently no preventative screen and is the fourth leading cause of cancer death in the United States. The high mortality is no doubt related to the lack of screening tools and therefore late diagnosis, by which time pancreatic adenocarcinoma is typically quite advanced with a paucity of effective treatments. With early diagnosis, surgical resection is a viable option. Therefore, plasma or stool testing are viable options, and a specific area in which cfDNA testing may be beneficial. Previous investigations have shown that mutant KRAS and methylated BMP3 can be detected in the stool with 90% specificity, but only ~50% sensitivity. 81 Several other genetic markers, specifically methylated EYA4, MDFI, and UCHL1, were tested for and found to be significantly increased in stool from patients with pancreatic adenocarcinoma but did not improve the area under the receiver operating characteristic curve. Undoubtedly, with further research a combination of cfDNA markers could be used to develop a routine screen for at risk individuals.
An advantage of stool or plasma cfDNA testing is that it assays the entire gastrointestinal tract and portions of the hepatobiliary system, whereas endoscopies and biopsies evaluate only the particular regions viewed/biopsied. Other strengths of cfDNA testing over current diagnostics are the ease of multiplexing multiple tests for multiple alleles, which can increase both sensitivity and specificity. Though there are current limits to how many cfDNA targets may be multiplexed in a single reaction vessel, there are no limits in the number of separate reactions that be performed in separate vessels for a gene or mutation panel approach to assaying. In addition, as the underlying technology is essentially the same regardless of the allele tested, this allows for rapid translation from the research benchtop to the clinical lab, and reduced expense as all DNA diagnostic technologies continue to fall in price. A reduction in price and ease of running tests could theoretically drive serial testing to monitor disease progression or treatment response,82–84 improve prognostic ability, and allow for rapid detection of treatment resistance so that modification to therapy can be done early.
Overall, we conclude that cfDNA tests can complement traditional gastroenterology and hepatology tools such as endoscopy, colonoscopy, ERCP, and traditional markers (such as plasma inflammatory markers, antibodies, and stool tests). The advantage of cfDNA testing relative to traditional diagnostics is sensitivity, versatility, ease of multiplexing, rapidly reducing costs, and noninvasive nature, which will allow for more frequent and quantitative testing.
Current problems
There are several technical challenges that must be overcome before cfDNA testing becomes widespread. First, there is no standard method of collecting circulating cfDNA, and the method of collection can have a major impact on whether there is cellular lysis resulting in dilution of cfDNA. Several studies have shown that lysis of immature red blood cells (reticulocytes) is a large source of dilution of cfDNA. Though most studies of cfDNA use plasma, others use serum. In addition, there are no standard conditions for the collection and storage of cfDNA samples, although commercial tubes exist that are geared towards reducing cell lysis for the purpose of cfDNA collection (StreckTM tubes). In addition, if not stored appropriately, for instance, without EDTA, endonucleases will rapidly degrade cfDNA. If cfDNA is purified, care must be taken that purification is not biased to certain types of fragments (i.e. larger fragments) when quantitative analysis follows.
In addition to these preanalytical issues with collection, processing, and storage, there are no standard protocols with regards to the analysis of cfDNA. As this technology matures, third-party validation of testing parameters must be taken into account such as the sensitivity, reliability, use of internal and external controls, and regulatory guidance from the FDA as well as laboratory regulatory bodies will be required. These challenges will also have to be balanced with the cost and clinical utility of performing cfDNA testing.
Other inherent problems with cfDNA include the small and fragmented nature, which makes analysis more difficult, and nearly impossible for the detection of mutations such as balanced chromosomal rearrangements.
Future directions
Regular use of cfDNA technologies in the clinical setting will not happen routinely until at least one application gains widespread acceptance as a useful, convenient and affordable tool by clinicians and patients. Noninvasive stool DNA testing for colon cancer may be this first common application that will trigger acceptance and routine use of these technologies, though no plasma testing is commonly used. Plasma testing is likely to begin in oncology before gaining traction in other medical specialties.
Once one plasma test becomes routine, the barrier to other cfDNA tests will fall as clinical laboratories obtain the equipment and expertise required for cfDNA testing. Once cfDNA sequencing, copy number, and methylation analysis of one allele can be performed routinely, the ability to perform the same test on other alleles will be much simpler, spurring further use. As an example, precursors to enzyme-linked immunosorbent assay (ELISA) were invented in the 1960s, and did not come into routine clinical use for some limited applications until the late 1970s, and it took until the 1980s for automated benchtop systems to become available. 85 It was after automated systems became available that testing for antibodies and antigens became routine enough that community practitioners started using this technology regularly.
The success of prenatal genetic testing may be a glimpse into the future potential of cfDNA testing in gastroenterology and hepatology. Noninvasive prenatal genetic testing is a cfDNA technology now widely used by pregnant women. These tests are extremely reliable with sensitivities and specificities for most abnormalities being on the order of 95.8–99.7% and ~99.86–99.96%, respectively. 86 This reliability is achieved even though it is estimated that fetal DNA concentrations in the maternal plasma are on the order of 1–6 ng/ml. 87 In fact, women undergoing in vitro fertilization are now able to have sequencing performed on a single cell harvested from blastomeres to determine which blastomeres to use prior to implantation. 88 In addition, proof-of-concept studies have shown that entire fetal genomes can be sequenced from fetal cfDNA in the maternal plasma.89,90 Taken together, the commercial and clinical success of noninvasive prenatal genetic testing demonstrates that it is clinically feasible with the ability to detect disease in small fractions of cfDNA and do so in a highly reliable fashion at acceptable cost. All of these excellent qualities could easily be translated into the diagnosis and management of digestive and liver diseases. As the technology itself matures, the main barrier to routine use will be research to develop markers with robust clinical applicability. cfDNA markers must add meaningfully to diagnosis, prognosis, and disease management. For instance, clinicians must be able to develop reliable algorithms about what to do with findings of high levels of mutant KRAS in the plasma of a patient with weight loss, nausea, and epigastric abdominal pain. For example, should such a patient undergo computed tomography (CT) imaging of the abdomen and pelvis, EGD, or positron emission tomography (PET)-CT? If these studies are negative, what are the next steps? Without preclinical and clinical research to guide clinicians, these types of dilemmas may add more to inability to obtain a diagnosis, cost, and anxiety rather than alleviate these problems. Therefore, to be beneficial results of cfDNA studies must be actionable.
Although these same challenges apply to the use of personalized medicine with genomics and germline DNA, one advantage of cfDNA over germline sequencing is the focus on tissue where the disease occurs. Whereas genomics helps to answer questions more geared towards disease risk over a lifetime and treatment options such as pharmacogenomics, cfDNA analysis is more focused towards changes to the genetics and epigenetics at a tissue level, similar to biopsy. This distinction has led to cfDNA analysis being termed a ‘liquid biopsy’ and similar to traditional biopsy is suited to diagnose disease or predisease states now, aid in treatment decisions (i.e. EGFR inhibitor versus BRAF inhibitor; glucocorticoids versus TNF inhibitor), and to monitor disease progression and treatment effectiveness.
In conclusion, we believe that as cfDNA analysis improves in sensitivity, specificity, cost, and ease of use, this technology will gain more traction in the preclinical realm as it evolves towards clinical use. The theoretical possibilities are enormous and this technology will undoubtedly become part of routine clinical use in gastroenterology and hepatology.