
Abstract
Background:
Gastrointestinal angiodysplasias (GIADs) could be responsible for recurrent bleeding and severe anemia. Somatostatin analogs could reduce transfusion requirements in these patients but no randomized controlled study is available. The main objective of the ANGIOPAS phase II double-blinded randomized, noncomparative study was to assess the effectiveness of pasireotide-LAR in reducing transfusion requirements in patients with refractory GIADs bleeding.
Methods:
A total of 22 patients with transfusion requirements ⩾6 units of packed red blood cells (pRBCs) during the 6 months prior to inclusion were randomized to receive pasireotide-LAR 60 mg (n = 10) or placebo (n = 12) every 28 days for 6 months. Patients were then followed for an additional 6 months after stopping treatment.
Results:
The pasireotide-LAR and placebo groups were equivalent for age, sex, comorbidities and transfusion requirement during the reference period (median 13 and 9.5 pRBCs). A 50 and 83% success rate (success defined as a decrease of at least 30% of transfused pRBCs) was observed in the pasireotide-LAR arm in the Intent to Treat (ITT) and per protocol (PP) analysis respectively. The need for transfusion during the intervention period was 3 pRBC units in the pasireotide-LAR group (range 0–26) and 11.5 pRBC units in the placebo group (range 0–23). Overall, three cases with glycemic control impairment were observed in the pasireotide-LAR group including one de novo diabetes.
Conclusion:
This double-blinded noncomparative randomized phase II study suggests, for the first time, the effectiveness of pasireotide-LAR 60 mg every 28 days to decrease the transfusion requirement in patients with recurrent bleeding due to GIADs.
Introduction
Gastrointestinal angiodysplasias (GIADs) are abnormal tortuous small blood vessels. GIADs are frequently multiple and preferentially observed in the proximal colon, the gastric antrum and the small bowel. 1 The prevalence of GIADs increases with age2–4 and presence of pathological conditions such as chronic renal failure, 5 liver cirrhosis, 6 aortic stenosis, 7 left ventricular assist devices 8 and von Willebrand’s disease. 9
GIADs may be responsible for occult or overt gastrointestinal bleeding. Despite frequent spontaneous bleeding stops, GIADs rebleeding is frequent.6,10–12 GIADs may require transfusion or endoscopic therapies using argon plasma coagulation or other techniques. In 20–40% of cases, GIADs rebleeding is observed despite optimal endoscopic therapy.13,14 In these situations, medical therapies are proposed. Hormonal therapy with estrogens and progesterone introduced in the 1950s are no longer used as they appear inefficient in a well conducted case-control study and a randomized study.15–18 Thalidomide 100 mg daily was reported to be useful in a series of patients and further proven to be efficient in a randomized study including relatively young patients.19–21 However, thalidomide’s frequent side effects limit its long-term use, especially in older patients. Few cases of patients with GIADs bleeding were recently treated with bevacizumab.22,23
Several observational studies and a meta-analysis consistently suggested that somatostatin analogs could reduce rebleeding and transfusion requirements in patients with GIADs bleeding refractory or inaccessible to endoscopic therapies24–44 (Table 1). However, the studies testing various therapeutic schedules used an inadequate design and failed to demonstrate this putative benefit. Indeed, no randomized study has properly evaluated somatostatin analogs in patients with GIADs bleeding refractory to endoscopic therapies.
Series and case-control studies evaluating somatostatin analogs effect in patients with GIADs rebleeding.

Hb, hemoglobin.
Pasireotide-LAR is a new long-acting analog of somatostatin. Like the natural somatostatin and other somatostatin analogs, pasireotide exerts its pharmacological activity via binding to the five somatostatin receptors subtypes. Pasireotide binds with high affinity to four of the five receptors with a specific 40-fold increased affinity to subtype five compared with other analogs. Pasireotide-LAR is mainly used for acromegaly and Cushing’s disease. The main objective of the ANGIOPAS study was to assess the effectiveness of pasireotide-LAR in reducing trans-fusion requirements in patients with GIADs recurrent bleeding refractory to endoscopic therapy.
Patients and methods
Study design
The ANGIOPAS study is a multicenter prospective double-blinded randomized noncomparative phase II study designed to evaluate the pasireotide-LAR interest in the control of GIADs refractory rebleeding. The study was divided into three periods: (1) preinclusion or basal period up to 6 months before randomization allowing the prospective selection of refractory GIADs patients with transfusion requirement ⩾6 units of pRBCs; (2) 6 months intervention period corresponding to the treatment period with intramuscular injection of pasireotide-LAR 60 mg or placebo every 28 days according to a 1:1 randomization and (3) follow-up period up to 6 months (Figure 1). The placebo consists of the same vehicle without the active drug (similar aspect and conditioning). The placebo was fabricated in the same facility as the tested drug. Both patients and investigators were blinded during the study. In case of active bleeding during the follow-up period, the patients could be treated with pasireotide-LAR in an unblinded manner. During intervention and follow-up periods, patients had monthly monitoring. At each visit, physical examination was performed and the following parameters were collected: age, weight, blood pressure, Karnofsky index, number of the pRBC units transfused, number of days of hospitalization for bleeding, endoscopic procedures, iron treatment. Blood sampling was performed monthly for hemoglobin, coagulation measurement, glycemia and liver function tests. Adverse events were collected at each visit. Electronic randomization and treatment allocation were performed by a central unit (CREAPHARM, Le Haillan, France). The ANGIOPAS study was performed according to the French and European regulations. The study protocol has been recorded on clinicalTrials.gov and European Clinical Trials web site under NCT02622906 and 2011-002579-40 number respectively. The ANGIOPAS study was approved by the Human Studies Committee at coordinator center PC/AP 31-2011. The permission for the study was obtained from Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) under the number AEC/A110808-42. This study was also approved by the Commission Nationale de l’Information et des Libertés (CNIL).
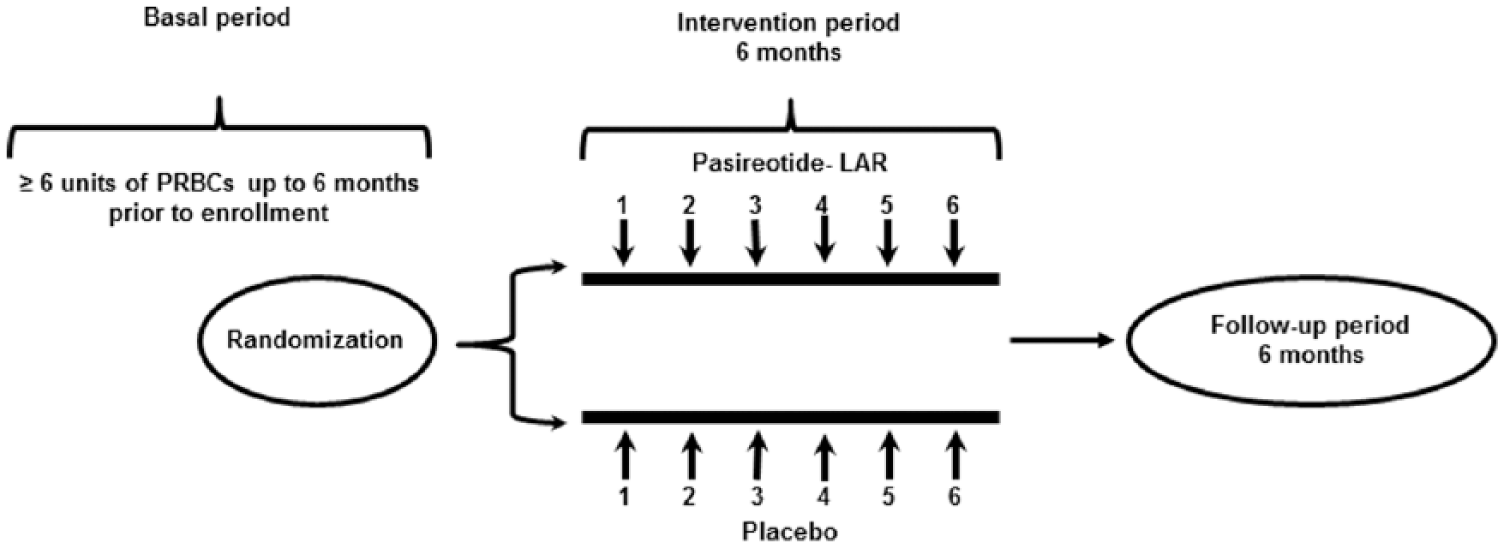
Study design.
Patient selection
All patients with uncontrolled occult or overt bleeding due to GIADs refractory or inaccessible to endoscopic therapies with transfusion requirement ⩾6 units of pRBCs from six tertiary hospital centers were considered for inclusion. Other inclusion criteria were age >18 years, presence of GIADs confirmed by endoscopy, failure or impossible endoscopic therapy, affiliation to the French national social security insurance and signed informed consent. Exclusion criteria were hereditary hemorrhagic telangiectasia, previous treatment with somatostatin analog during the 6 months prior to inclusion, symptomatic cholelithiasis, uncontrolled diabetes [glycated hemoglobin (HbA1c) >8%], previous bleeding from esophageal varices, aspartame amino-transferase (ASAT) or alanine amino-transferase (ALAT) > 2 upper limit of normal (ULN) or total bilirubin > 1.5 ULN, prothrombin (TP) < 50%, platelets < 75,000/mm3, activated partial thromboplastin time (aPTT) > 1.5-times the control, QTcF > 450 ms, history of syncope, history of ventricular tachycardia or fibrillation, familial history of idiopathic sudden death, myocardial infarction within 6 months, congestive heart failure grade III and New York Heart Association, metastatic malignancy and impossible follow up for psychological or geographical reasons. Patients with antivitamin K therapies were allowed to participate using cautious deep subcutaneous injection. Pregnant or nursing women, women and men of reproductive age without effective contraception and women of childbearing age who have not carried out a pregnancy test were not allowed to participate.
Criteria of evaluation
The main evaluated outcome was the evolution in transfusion requirement during the 6 months treatment and follow-up periods. These time periods were selected according to available data from observational studies suggesting that a preventive effect could be demonstrated when such a period is considered. For the analysis of this primary endpoint, success was arbitrarily defined as a minimum 30% reduction in the number of transfused pRBCs between the 6-month period prior to randomization and 6 months after randomization. A 30% decrease in transfusion requirement was arbitrarily selected according to expected effect from observational studies. This quantity of effect was also considered since such a decrease could convince of the real interest of this therapy. Pasireotide-LAR was arbitrary classified as of no or futile interest for a proportion of success <15% and of interest for future phase III studies for a proportion of success ⩾40%. The secondary outcomes were the evolution in the number of transfused pRBCs, the number of patients requiring transfusion, the number of endoscopic procedures and the number and days of hospitalization. Assessment of toxicity was performed during monthly visits including physical examination and blood testing.
Statistical analysis
Patient data were compared between basal and treatment period for each arm. A one-stage Fleming–A’Hern design was used to evaluate the probability that p ⩽ p0 and p ⩾ p1 where p is the observed proportion of success, p0, chosen as 15%, was the maximal expected proportion of success for a drug with no or futile effectiveness and p1, chosen as 40%, was the minimal expected proportion of success for a truly effective drug requiring future phase III studies. According to these hypotheses, the number needed to be included in the treated group was calculated to be 22 for a 0.05 α bilateral risk and a 0.80 β risk. If at least 7 out of 22 patients meet the response criteria, pasireotide-LAR will be considered effective. The recruitment of a control placebo group of 22 patients was also planned to better describe the natural history of these patients and reduce patient selection bias. As stated previously, no statistical comparison was originally scheduled between the two arms in this noncomparative phase II study. So the study size was not calculated to demonstrate a difference between the pasireotide-LAR and placebo groups.
The primary analysis was performed by Intent to Treat (ITT) for all randomized patients regardless whether treatment was received or not. In the ITT analysis, the worst possible value for the primary endpoint was attributed to missing data. So a case with incomplete treatment in the treated arm was classified as failure whatever the observed result. Whereas, a case in the placebo group with incomplete treatment was classified as a success, whatever the observed result. In addition, an explanatory per protocol (PP) analysis of all patients randomized who received all planned injections and treated without other violation or major deviation from protocol was performed.
Wilcoxon’s test was performed to compare the transfusion requirement between the reference period and intervention period for each arm. The 95% confidence intervals (CIs) were calculated. A p-value < 0.05 was considered statistically significant. Analyses were carried out using R statistical software version 3.1.2 (R Foundation for Statistical Computing, Vienna, Austria, http://www.r-project.org). All authors had access to the study data and reviewed and approved the final manuscript.
Results
Recruitment
From March 2012 to December 2013, 25 consecutive eligible patients from six tertiary hospital centers were considered for inclusion. A total of 22 patients (16 men and 6 women) were randomized, 10 in the pasireotide-LAR group and 12 in the placebo group (Figure 2). The planned additional 6 months of follow up without treatment was complete in four patients in the pasireotide-LAR group and in four in the placebo group. The pasireotide-LAR and placebo groups were equivalent for age (mean 70 ± 8 and 72 ± 16 years respectively), sex (7 and 9 men), weight (73 ± 15 and 72 ± 14 kg), transfusion requirements during the reference basal period (median 13 and 9.5 pRBCs) and hemoglobin at baseline (mean 9.6 ± 1.2 and 9.2 ± 1.9 g/dl) (Table 2). The median duration of bleeding before randomization was 36 months (range 3–192). Melena and rectal bleeding were observed at inclusion in nine and four cases respectively. All patients had at least an upper endoscopy, a colonoscopy and a small bowel capsule endoscopy. Enteroscopy with argon plasma coagulation was performed before inclusion in 21 cases (1–5 sessions). All patients had multiple GIADs. The most frequent GIAD localization was the small bowel (n = 21), then the proximal colon (n = 11) and the stomach (n = 7). A total of 12 patients out of the 22 (55%) had multiple GIADs localization. Comorbidities including ischemic disease requiring or not stenting and valvulopathy with or without valve replacement were frequent in both groups (Table 2). Concomitant medication with anticoagulants or antiplatelet agents was present at inclusion in 13 cases (60%) (Table 2).

Study flow chart.
Demographic data.

BMI, body mass index; Hb, hemoglobin; GIAD, gastrointestinal angiodysplasia; pRBC, packed red blood cell; SD, standard deviation.
Primary endpoint
Pasireotide-LAR group
In the ITT primary analysis, according to the method of the worst situation, four patients who did not receive the full 6 months of the scheduled intervention were arbitrarily classified as failures regardless of the evolution in transfusion requirement. With this restrictive method, success was characterized in 5 out of the 10 patients (50%; 95% CI 19–81%) in the pasireotide-LAR group. In the PP analysis, without this restrictive analysis, the success rate was 83% (95% CI 36–99%) (Table 3).
Endpoints.

CI, confidence interval; pRBC, packed red blood cell.
Overall, two patients from the placebo group with persistent bleeding after the placebo 6 months treatment were treated with pasireotide-LAR in an unblinded manner. According to the analysis of these 12 patients (10 patients of the pasireotide-LAR group and 2 patients of the placebo group) treated with pasireotide-LAR, the ITT success rate was 58% (95% CI 28–85%) and the PP success rate was 88% (95% CI 47–99%).
Suspension of the pasireotide-LAR 6 months treatment in two patients from the pasireotide-LAR group with no further bleeding during treatment was followed by renewed bleeding. In one of these two cases, resumption of pasireotide-LAR led to a further bleeding stop.
Placebo group
Mirroring and using the described worst case method, 4 patients out of the 12 in the placebo group who did not receive the full 6 months of placebo were arbitrarily classified as successes regardless of the evolution in transfusion requirement. Then, ITT success rate in the placebo arm was 50% (95% CI 21–79%). This rate is the same as in the pasireotide arm according to the method of the worst situation yet presented. When this artificial method was not used, the observed PP success rate was 25% (95% CI 3–65%).
Secondary endpoints
The median number of transfused pRBC units during the treatment period was 3 (range 0–26) in the pasireotide-LAR group and 11.5 (range 0–23) in the placebo group. The evolution of individual pRBC requirements is presented in Figure 3. The delta of pRBCs between the pasireotide-LAR treatment period and the reference period was −67% (p = 0.035). This delta was +18% in the placebo group (p = 0.94). Spontaneous stop bleeding was observed in two out of eight patients in the placebo group: 22 and 15 pRBC transfusion requirements during basal period versus 0 and 2 units during 6 months of follow up. No differences were observed in the proportion of patients requiring transfusion (67% in the pasireotide-LAR group and 75% in the placebo group), the proportion of patients requiring new endoscopy (17 and 25%) nor the proportion of patients requiring new hospitalization (83 and 63%). Exploratory analysis did not suggest any difference in these effects according to age (>70 or >70 years) nor to concomitant therapy with or without anticoagulant or antiplatelet (see Table 4).
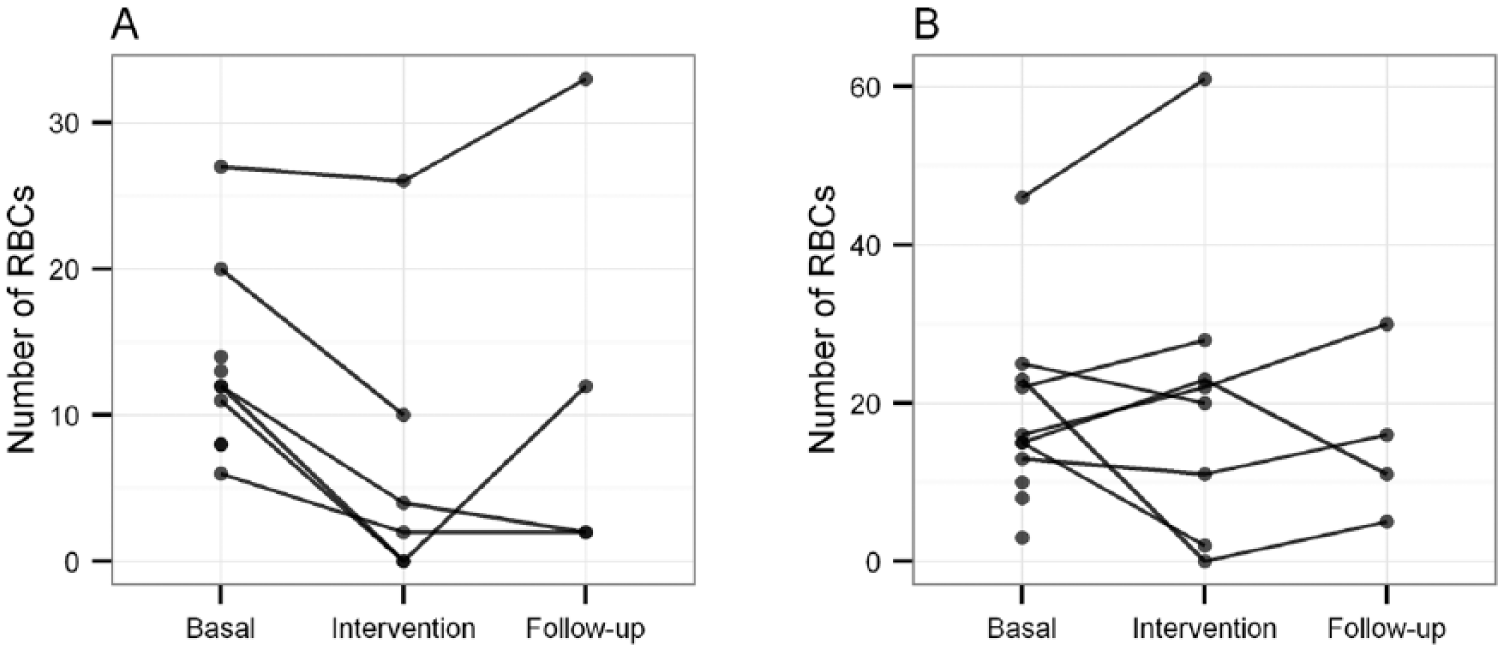
Evolution of pRBCs transfusion requirement in pasireotide-LAR and placebo treated patients.
Exploratory analysis.

ITT, Intent to Treat; PP, per protocol.
Safety
Overall, two deaths occurred during the study: one in the placebo group and one in the treated group and considered unrelated to pasireotide (Table 5). This death occurred in a context of worsening condition in a patient with noninsulin-dependent diabetes, cardiomyopathy, chronic obstructive pulmonary disease, chronic liver disease and pulmonary hypertension. By the end of 2015, three additional deaths were observed, two in the placebo group and one in the pasireotide-LAR group which were considered unrelated to the treatment.
Adverse events.

Overall, three cases with deterioration in glycemic control were observed in the pasireotide-LAR group including one with de novo diabetes requiring insulin therapy without need for PP treatment discontinuation. In addition, the diabetes treatment had to be adapted in a patient. Hypoglycemia associated with diarrhea was observed in one patient several days after the first pasireotide-LAR injection. This patient had a history of insulin-dependent diabetes, dyslipidemia, hypertension and peripheral arterial disease. After the onset of this adverse event, the investigator decided to stop the use of the product. The patient was monitored until complete disappearance of the adverse event few days later. A total of three cases of mild diarrhea were observed in the pasireotide-LAR group. Grade 1 transient nausea requiring no specific treatment was reported in one patient in the pasireotide-LAR group. No symptomatic biliary event was reported. No unexpected adverse events were observed during the study.
Discussion
Despite the use of a worst case scenario analysis, a 50% success rate in reducing the transfusion requirement was observed in the pasireotide-LAR arm (n = 10, ITT analysis). The success rate was arbitrarily defined as a decrease of more than 30% of transfusion requirements between the treated and the basal period in patients requiring at least 6 pRBC units during the last 6 months. We considered this threshold to be clinically relevant in such a population. In this ITT analysis, the worst possible value that is failure was attributed in case of true failure but also in case of incomplete follow up, so this 50% success rate is a very conservative rate. Indeed, the success rate was 83% in the PP analysis of these 10 patients and 88% when all 12 pasireotide-LAR-treated patients were considered. As the PP success rate was lower in the placebo group and since patients had a long history of recurrent bleeding (median 36 months), we speculate that this effect was not due to spontaneous bleeding stop but to true pasireotide-LAR protection. This protective effect appears clearly in the changes in pRBC transfusion requirements between treatment and the reference period in the pasireotide-LAR arm (−67%).
These results are in line with previous observational data indicating a decrease in bleeding and pRBC transfusion requirements in patients treated with octreotide, octreotide-LAR or lanreotide.24–43 The quantity of effect observed in the ANGIOPAS study was in the range of these cases and in particular, of three series with significant transfusion requirements.39,40,43
We did not investigate the mechanism of action of pasireotide-LAR in this study. These putative mechanisms were described in an original paper 45 and in recent in-depth reviews.46,47 The discussed mechanisms involved both physical factors with increased local tension and abnormal angiogenesis due to hypoxia as well as acquired hemostasis. In this study, the population was relatively old with cardiovascular comorbidities including ischemia and aortic valvular disease suggesting a putative role of ischemia. Patients were also frequently exposed to anticoagulants or antiplatelet agents, key contributing factors to rebleeding.
The main limitation of this study is its limited recruitment. The initial recruitment ANGIOPAS target has not been achieved. Only 25 patients were selected and 22 included despite 50 were initially considered. Several factors may explain this limited recruitment: scarcity of the pathology at the desired stage (refractory bleeding after endoscopic therapy with transfusion requirement of more than 6 pRBC units during the last 6 months), exclusion criteria as QTcF > 450 ms which was frequently observed in these elderly patients with vascular comorbidities and the geographical remoteness of some patients that do not allow regular monitoring according to the protocol. This difficulty was also described by another team. 41 Despite this limited recruitment and the restrictive attribution method for missing data, the observed success rates allow the rejection of a futile effect (p < p0 hypothesis when p0 < 15% with a 0.037 true error rate). Furthermore, the PP analysis of the patients exposed to pasireotide-LAR suggests the true effectiveness of pasireotide-LAR in reducing the transfusion requirement (p ⩾ p1 hypothesis with p1 ⩾ 40%).
The balance between benefit and potential harm needs to be better evaluated in larger series but according to the refractory severe bleeding condition and fragile status of patients, a favorable balance is expected.
These results suggest effectiveness of pasireotide-LAR in the control of recurrent bleeding due to refractory GIADs. A multicenter collaborative network involving specialized centers will allow performing the needed phase III study to confirm these results.
Footnotes
Acknowledgements
This work was promoted by the SFED (Société Française d’Endoscopie Digestive) and was conducted by his scientific committee. We thank Dr Bisot-Locard for her full commitment in this study. This work is listed under ClinicalTrials.gov identifier: NCT02622906.
Funding
This research received funding from Novartis laboratory. Novartis laboratory did not participate in the elaboration of the study design, protocol or manuscript writing.
Conflict of interest statement
RB: Alfa Wassermann Pharma, Mayoly Spindler, Medtronic and Norgine.
MoB: None.
JCS: Abbvie, Bouchara, Intromedic, Mayoly Spindler, Medtronic, MSD and Recordati.
ChC: Abbvie, Aptalis, Gilead, MSD, Sciences Janssen and Vifor Pharma.
RL: Abbvie and Boston Scientific.
MV: None.
CB: Boston Scientific, Cousin Endosurg, Norgine and Olympus.
MaB: None.
RG: Ipsen Pharma.
ES: Amgen, Bayer, Ipsen Pharma, Novartis Oncology, Pierre Fabre, Pfizer, Roche, Sanofi and Vifor Pharma.
PG: Abbvie, Boston Scientific, Cook Medical, Mayoly Spindler and Takeda.
TA: Amgen, Novartis Oncology, Pfizer, Roche, Sanofi and Vifor Pharma.
GA: None.
BB: None.
CB: None.
JJR: None.
VL: Amgen, GSK, Mundi Pharma, Octa Pharma and Roche
DS: Boston Scientific.