
Abstract
Irritable bowel syndrome (IBS) is a common functional gastrointestinal disorder characterized by abdominal pain associated with defecation with altered stool frequency or stool form. The global prevalence of IBS ranges from 10% to 15% and total healthcare cost attributable to IBS is significant. Among individuals with IBS, the condition has dramatic effects on health-related quality of life, work and school productivity, and activities of daily living. It may be diagnosed with confidence, based on symptom-based diagnostic criteria, exclusion of alarm features and directed diagnostic testing. Management of IBS typically begins with dietary and lifestyle modifications, progressing to over-the-counter therapies, and then to prescription medications, both approved and nonapproved for IBS. This narrative summarizes the efficacy and safety of three US Food and Drug Administration (FDA)-approved prescription therapies for IBS with diarrhea (IBS-D), with a focus on the most recently marketed agent, eluxadoline, and its role in the treatment IBS-D.
Introduction
Irritable bowel syndrome (IBS) is the most common functional gastrointestinal (GI) disorder diagnosed worldwide. The prevalence of IBS varies across geographical and socioeconomic environments, highlighting the heterogeneity of the condition, both with respect to etiologies and clinical presentation. 1 The global prevalence of IBS is estimated to be 10–15%.2–5 IBS is defined by the Rome IV criteria as the presence of abdominal pain occurring at least once a week over the past 3 months, associated with altered stool frequency or form. Symptom onset should occur at least 6 months prior to the diagnosis. Other symptoms commonly reported by patients with IBS include bloating, abdominal distension, abdominal cramps, flatulence, fecal urgency, and fecal incontinence. 2 IBS can be categorized into three primary subgroups based on the predominant bowel complaint: IBS with diarrhea (IBS-D), IBS with constipation (IBS-C), and IBS with a mixed stool pattern (IBS-M).
The pathogenesis of IBS is thought to be multifactorial, involving visceral hypersensitivity, dysregulation of the brain–gut axis, abnormal gut motility, absorption and secretion, altered immune function and psychosocial distress. 2 In addition, alterations in the gut microbiota may play a role in the pathogenesis of IBS. Genetic and environmental factors, such as diet, infections and antibiotics have been proposed as mechanisms for dysbiosis and IBS symptoms. 1 Rather than a single disease state, it is becoming increasingly clear that the varied etiologies enumerated above (as well as others that are less proven) can act alone or in concert to produce the characteristic clinical symptoms that comprise IBS.
Regardless of the underlying pathophysiology, IBS has a significant negative impact on quality of life of affected individuals and greatly reduces their work productivity. Excessive physician visits, diagnostic testing, and prescription and over-the-counter (OTC) medications contribute to the high healthcare costs attributable to this condition. It is estimated that IBS patients account for 3.6 million physician office visits annually with total healthcare costs estimated to be greater than $30 billion per year. 2 The management of IBS is typically tiered and directed at the most bothersome symptoms, beginning with lifestyle modifications then OTC therapies and then prescription therapies. Such a symptom-directed therapeutic approach often involves multiple medication trials and fails to address the underlying pathogenesis of this complex disorder. This review will summarize the clinical evidence for three approved prescription therapies for IBS-D, with a specific focus on the clinical development program of eluxadoline in order to describe its place among the different management approaches for IBS-D.
Alosetron
Alosetron is a selective serotonin 5HT-3 antagonist indicated for the treatment of women with severe IBS-D who have failed conventional therapy. 5HT-3 receptors are distributed on enteric neurons of the gastrointestinal tract, as well as throughout the peripheral and central nervous systems. Mechanistically, alosetron alters gastrointestinal secretion, slows colonic transit, increases colonic compliance, and alters the perception of visceral pain. 6
As part of its clinical development and approval process, two double-blinded, randomized, placebo-controlled trials for alosetron were performed in women with IBS-D or IBS-M in which alosetron 1 mg twice daily (BID) was compared with placebo BID for a total of 12 weeks. In both clinical trials, alosetron provided adequate relief of IBS-related abdominal pain, the primary endpoint, in significantly more patients for all 3 months of treatment compared with placebo; 43% versus 26% [difference 17%, 95% confidence interval (CI): 8.0–25.4] in one trial and 41% versus 29% (difference 12%, 95% CI: 4.7–19.2) in the other.6,7 In addition, secondary endpoints, including a decreased sense of urgency, decreased stool frequency, and increased stool firmness were significantly improved with alosetron versus placebo.
Constipation was the most common drug-related adverse event (AE) in the two trials and occurred most frequently in the alosetron-treated groups; 25% versus 5% with placebo 7 and 30% versus 3% with placebo, 6 respectively. No drug-related serious adverse events (SAEs) or deaths were reported in either study. There was one reported case of colitis, however, histological and immunohistochemical staining confirmed an infectious etiology. 6 Prior to US Food and Drug Administration (FDA) approval, the safety profile of alosetron accumulated from preclinical and clinical trials identified four known cases of colon ischemia and dose-dependent constipation. 8 However, shortly after its approval, numerous reports of colon ischemia and complicated constipation were reported. 9 Alosetron was eventually withdrawn from the market in November 2000, after several deaths were associated with its use. After aggressive lobbying, alosetron was reintroduced to the market in November 2002, accompanied by a lower initiating dose (0.5 mg BID) and restricted indication for use along with an FDA-monitored risk management program (RMP).8,10 Over the last several years, the RMP for alosetron has been significantly relaxed. While demonstration of provider understanding of the restricted indication and risks associated with alosetron is still required, placement of RMP enrollment stickers on handwritten prescriptions and obtaining patient signatures on an RMP specific ‘consent form’ are no longer mandated. A recent review of 9 years of postmarketing safety data for alosetron reported an infrequent and stable adjudicated incidence rate of colon ischemia (1.03 cases/1000 patient-years) and a low and declining adjudicated incidence rate of complications of constipation (0.25 cases/1000 patient-years). 11
Rifaximin
Rifaximin is approved for adult men and women with IBS-D. It is an oral, nonsystemic, locally acting rifamycin-derived antibiotic that has been shown to inhibit bacterial protein synthesis. While the exact mechanism of action of rifaximin for IBS-D is not known, it is thought that it primarily exerts its clinical effects via alterations in the gut microbiota. 12
The efficacy and safety of rifaximin for IBS were assessed in the TARGET 1 and TARGET 2 trials, two identical phase III placebo-controlled clinical trials in which patients with non-IBS-C were randomized to receive either rifaximin 550 mg three times daily (TID) or placebo TID for 14 days. Patients were followed for an additional 10 weeks post-treatment. The primary endpoint of these trials was adequate relief of global IBS symptoms for at least 2 of the 4 weeks after completion of therapy. The onset and duration of therapeutic effect was also assessed by determining the monthly response rate, defined as the proportion of patients with adequate relief of global IBS symptoms for at least 2 weeks per month during months 1–3. Relief of IBS-related bloating was a key secondary endpoint. 12 In both studies, significantly more patients treated with rifaximin achieved adequate relief of global IBS symptoms compared with placebo (40.8% versus 31.2%, p = 0.01, TARGET 1 and 40.6% versus 32.3%, p = 0.03, TARGET 2). Adequate relief of global symptoms within the first month, with continued relief during the first 2 months, occurred more frequently in the rifaximin-treated group compared with placebo [odds ratio (OR) 1.35, 95% CI: 1.0–1.82; p = 0.05, TARGET 1 and OR 1.52, 95% CI: 1.13–2.03, p = 0.005, TARGET 2]. In addition, adequate relief of IBS-related bloating occurred in significantly more patients treated with rifaximin compared with placebo (39.5% versus 28.7%, p = 0.005, TARGET 1 and 41% versus 31.9%, p = 0.02, TARGET 2). 12
TARGET 3 was a third phase III clinical trial designed to address concerns of the FDA regarding the efficacy, safety, and potential emergence of resistant bacterial strains with multiple treatments with rifaximin in IBS-D patients who initially experienced a therapeutic response to the medication, but subsequently suffered a relapse of symptoms. 13 All enrolled patients were initially treated in a blinded fashion with placebo, and those who failed to respond to placebo during this ‘precebo’ phase were treated with open-label rifaximin 550 mg TID for 14 days. Responders were defined as those who experienced a simultaneous ⩾30% decrease from baseline mean abdominal pain score and ⩾50% decrease from baseline in the number of days per week with Bristol Stool Form Scale type 6 or 7 for at least 2 of the 4 weeks post-treatment. Nonresponders from the open-label phase of the trial were withdrawn from the study. Responders, however, were monitored for up to 18 weeks or until relapse of their IBS-D symptoms occurred. Patients who experienced a relapse in IBS-D symptoms were then randomized (1:1) to received rifaximin 550 mg TID or placebo TID for 14 days. This represented the primary evaluation period for this trial. The primary endpoint of this complicated trial was the proportion of patients with recurrent IBS-D symptoms who met responder criteria for at least 2 of the 4 weeks that immediately followed the first repeat treatment. A second repeat treatment was included to assess the safety of multiple courses of rifaximin and to evaluate for the emergence of bacterial resistance. In terms of results, 44% of IBS-D patients met the responder definition after open-label rifaximin. Among these responders, approximately two-thirds experienced recurrent IBS-D symptoms during 18 weeks of observation, with a mean time to recurrence of 10 weeks. After the first retreatment, statistically significantly more subjects retreated with rifaximin met responder criteria compared with placebo (38% versus 31%, p < 0.05).
Rifaximin has a favorable safety profile. In both TARGET 1 and TARGET 2, there were no clinically significant differences between rifaximin and placebo-treated patients for the incidence of AEs and SAEs. There were no reported cases of Clostridium difficile-associated diarrhea, colon ischemia or deaths. 12 Likewise, there was no significant difference in the frequency of clinically significant AEs or SAEs between groups during the TARGET 3 trial. There was a single case of C. difficile colitis reported in a patient after repeat rifaximin treatment. The patient had a past medical history of C. difficile infection and developed C. difficile colitis immediately after completing a 10-day course of a cephalosporin for a urinary tract infection. 13
Eluxadoline
Mechanism of action
Eluxadoline is an orally administered, locally acting, mixed opioid-receptor modulator 14 approved for the treatment of IBS-D in adult men and women. In the gut, there are three main opioid receptors, mu (µ), delta (δ), and kappa (κ), all of which are G-protein-coupled receptors. 15 These receptors are present in the enteric nervous system, smooth muscle cells, and interstitial cells of Cajal (ICC) where they are believed to play a major role in gut motility, secretion and visceral sensation. Eluxadoline is a µ-opioid and κ-opioid-receptor agonist/δ-opioid-receptor antagonist (Figure 1). It has been demonstrated that local activation of µ-opioid receptors slows GI transit and reduces GI secretions. 15 In contrast, δ-opioid receptor antagonism is thought to increase colonic propulsion. Thus, the addition of a δ-opioid-receptor antagonist to a µ-opioid-receptor agonist should theoretically counteract excessive inhibition of gastrointestinal motility of unopposed µ-opioid-receptor agonism, resulting in potentially more normalized bowel function. The concept of δ-opioid-receptor antagonism opposing µ-opioid-receptor agonism was demonstrated by Wade et al. using a murine model. 14 Coadministration of naltrindole (δ-opioid-receptor antagonist) with loperamide or morphine (µ-opioid-receptor agonists) reduced the extent of inhibited GI transit time compared with administration of either loperamide or morphine alone. There is also evidence to suggest that the combination of µ-opioid-receptor agonism and δ-opioid-receptor antagonism has synergistic analgesic effects. 15 The role of κ-opioid agonism is not well established, however, it may be involved in reducing visceral hyperalgesia.
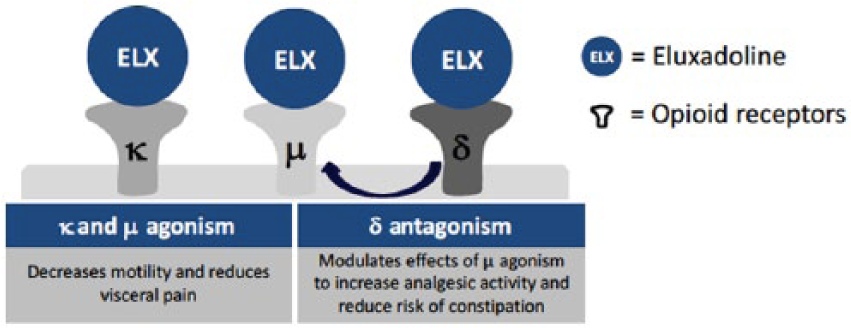
Mechanism of action of Eluxadoline ©Allergan, 2015. Used with permission from Allergan.
Preclinical trials assessing the opioid-receptor modulatory effects of eluxadoline on GI function utilized murine models. Mice were exposed to environmental stress resulting in a two-to-threefold increase in 1-hour fecal output. 14 Eluxadoline reduced fecal output to within control levels over a wide dose range without significantly suppressing fecal output below that of controls. In contrast, loperamide significantly inhibited fecal output below control levels, in essence, constipating the mice. Eluxadoline reduced gut transit in a dose-dependent fashion, however, it did not completely suppress transit even at high doses (50 and 100 mg/kg). By comparison, loperamide at a dose of 10 mg/kg completely abolished GI transit. These findings suggest that eluxadoline can normalize gut motility with less risk of constipation compared with loperamide. In isolated mice intestines, fluid and ion transport were induced by electrical field stimulation. There was no change in secretion seen with loperamide, however, eluxadoline inhibited the secretory response in the small bowel and colon. Eluxadoline also demonstrated an analgesic effect in a rat model of visceral hyperalgesia induced by repetitive intracolonic balloon distension. 14
Efficacy
The efficacy of eluxadoline in the treatment of IBS-D was evaluated in a phase II and two phase III randomized, double-blinded, placebo-controlled clinical trials.16,17 In the phase II trial, men and women who met Rome III criteria for IBS-D and who reported a mean worst abdominal pain (WAP) score of ⩾3, as well as a mean daily stool consistency score of ⩾5.5 on the Bristol Stool Form Scale (BSFS) were eligible for participation. 16 Patients were randomized to receive either placebo or eluxadoline 5, 25, 100, or 200 mg BID for 12 weeks. Follow-up visits were scheduled at weeks 2, 4, 8, and 12, with a subsequent 2-week post-treatment assessment at week 14. The primary endpoint was the proportion of patients who achieved clinical response at week 4. Clinical response was defined as a reduction in the mean WAP score by ⩾30% from baseline and by at least 2 points, and a daily BSFS of 3 or 4 on ⩾66% of days during week 4. There were multiple secondary endpoints, including clinical response at week 12, adequate relief of IBS symptoms, bowel movement frequency, urgency, IBS global symptom score, IBS severity symptom score, and quality of life, based on the IBS Quality of Life questionnaire (IBS-QOL). The primary endpoint was achieved by significantly more patients treated with 200 mg of eluxadoline compared with placebo (13.8% versus 5.7%, p < 0.05). Although statistical significance was not achieved at week 4 for the 100 mg eluxadoline group, a trend toward improvement was seen over placebo (11% versus 5.7%, p = 0.09). At week 12, significantly more patients receiving 100 mg of eluxadoline achieved clinical response compared with placebo (20.2% versus 11.3%, p < 0.05), however the proportion of clinical responders was similar among the 200 mg eluxadoline group and placebo. Patients treated with 100 or 200 mg of eluxadoline were more likely to report adequate relief of IBS symptoms compared with placebo at weeks 4, 8, and 12. In addition, patients treated with 100 or 200 mg of eluxadoline had greater improvements in bowel movement frequency, urgency, IBS global symptom scores, IBS symptom severity scores and quality-of-life scores compared with placebo (p < 0.05). A post hoc analysis was conducted using the FDA responder definition of ⩾30% reduction from baseline of WAP score and a simultaneous BSFS < 5 or no bowel movement on at least 50% of days during the 12 weeks. Both the 100 mg (28%) and 200 mg (28.5%) eluxadoline groups met the FDA responder endpoint significantly more often than placebo (13.8%, p < 0.005).
While eluxadoline at both doses evaluated in the phase II trial resulted in the reduction of pain and diarrhea in IBS-D patients, the 200 mg BID dose was associated with higher rates of AEs, so the 100 and 75 mg BID doses were chosen for the two phase III clinical trials. 17 Adult men and women who met Rome III criteria for IBS-D and who had a mean WAP score > 3, mean BSFS ⩾ 5.5, a BSFS ⩾ 5 for at least 5 days, and a mean IBS-D global symptom score of ⩾2 were eligible to participate. Patients were randomized to receive either eluxadoline at a dose of 100 or 75 mg BID or placebo for 26 weeks. In one of these studies, IBS-3001, participants received an additional 26 weeks of treatment for the collection of drug safety data, with a subsequent 2-week post-treatment follow-up period. In the other phase III trial, IBS-3002, the 26-week treatment period was followed by a 4-week single-blinded placebo withdrawal period, during which all patients received placebo to assess for rebound worsening of symptoms. The primary endpoint was the proportion of patients who had a composite response of simultaneous daily improvement in both abdominal pain (reduction of ⩾30% WAP score from baseline) and stool consistency (BSFS < 5 or absence of a bowel movement) on at least 50% of days over weeks 1–12 (FDA endpoint) and weeks 1–26 [European Medicines Agency (EMA) endpoint]. Secondary endpoints included abdominal pain relief (⩾30% reduction in WAP on ⩾50% of days), improvement in stool consistency (BSFS < 5 or absence of a bowel movement if ⩾30% reduction in WAP on ⩾50% of days), adequate relief of IBS symptoms, improvement in IBS global symptom score and improvement in IBS-QOL questionnaire score. The intention-to-treat population included 1280 patients in IBS-3001 and 1145 patients in IBS-3002. In both trials, the FDA primary efficacy endpoint was met by significantly more patients who received eluxadoline, either the 75 or 100 mg BID doses, compared with placebo (23.9% and 25.1% versus 17.1%, p = 0.01 and p = 0.004, respectively, in IBS-3001 and 28.9% and 29.6% versus 16.2% p < 0.001 in IBS-3002). Similarly, the proportion of patients who met the EMA primary efficacy endpoint was significantly higher in the 100 mg eluxadoline group compared with placebo in both clinical trials (29.3% versus 19%, p < 0.001 in IBS-3001 and 32.7% versus 20.2%, p < 0.001 in IBS-3002). Although there were more EMA responders in the 75 mg eluxadoline group compared with placebo in IBS-3002 (30.4% versus 20.2%, p < 0.001), there was no significant difference found between those who received 75 mg of eluxadoline BID and those who received placebo in IBS-3001 (23.4 versus 19%, p = 0.11). In addition, both trials found that treatment with eluxadoline (either 75 or 100 mg BID) resulted in a significant improvement over placebo in stool consistency, adequate relief of IBS symptoms and improvements in IBS global symptom scores and IBS-QOL questionnaire scores. The secondary endpoint of abdominal pain relief (⩾30% reduction in WAP on ⩾50% of days) was not statistically significant for either dose of eluxadoline, however, a pooled data analysis demonstrated a significant improvement in abdominal pain when defined by a ⩾40% or ⩾50% reduction in WAP from baseline for 100 mg of eluxadoline BID during weeks 1–12 and 1–26, and for 75 mg of eluxadoline during weeks 1–12.
Safety
A recent article reviewed the safety of eluxadoline throughout its clinical trial development period. 18 In general, the most common AEs reported in eluxadoline-treated patients were abdominal pain, nausea, vomiting, constipation and headache, occurring in ⩾2% of patients. These AEs occurred more frequently in eluxadoline-treated patients compared with placebo. Similarly, the proportion of patients with an AE resulting in study drug discontinuation was higher in the eluxadoline-treated groups compared with placebo, however there was no difference between eluxadoline doses (8.3% with 75 mg, 7.8% with 100 mg versus 4.3% with placebo). The overall frequency of SAEs was low, with a slightly higher frequency in the eluxadoline-treated groups compared with placebo (4.2% with 75 mg, 4% with 100 mg versus 2.6% with placebo). The most commonly reported SAE in the eluxadoline-treated patients was pancreatitis (0.4% with 75 mg, 0.4% with 100 mg), however, the overall incidence remained low (0.4%). There was a single case of colon ischemia that occurred in the 100 mg eluxadoline group and the patient recovered without sequela. There were no drug-related deaths. Patients without a gallbladder did have an increased incidence of AEs and SAEs across all treatment groups, including non-GI-related events. Both AEs and SAEs were most frequently reported within the first 2 weeks of study drug initiation for those patients treated with eluxadoline. The most common SAE reported during this time was pancreatitis (0.2% with 100 mg). By 12 weeks of treatment, the incidence of AEs was not different across all treatment groups (49.3% with 75 mg, 44.3% with 100 mg, 42.4% with placebo), although the incidence of SAEs was slightly higher in the eluxadoline-treated patients compared with placebo (2.5% with 75 mg, 2% with 100 mg versus 1.3% with placebo). Similarly, at 26 weeks of treatment, the incidence of AEs and SAEs were similar across treatment groups, however, the 100 mg eluxadoline group had a slightly higher incidence of SAEs.
The overall incidence of constipation with eluxadoline in clinical trials was low, with the majority of cases reported within the first 3 months of drug initiation. 18 Although the incidence of constipation was lower in the placebo group (2.5%) compared with the eluxadoline-treated groups, there was no significant difference in the incidence of constipation between the two eluxadoline doses (8.1% with 100 mg and 7.4% with 75 mg). Over the course of the clinical trials, the incidence of constipation declined over time and were similar across all treatment groups during weeks 4–12. There were no SAEs of constipation reported in any treatment groups. As would be expected, eluxadoline is contraindicated in patients with chronic or severe constipation or mechanical gastrointestinal obstruction.
Opiates are known risk factors for the development of sphincter of Oddi spasm (SOS) and pancreatitis, likely due to their direct action on the sphincter of Oddi (SO). 19 µ-opioid-receptor agonism increases the amplitude and frequency of SO phasic wave contractions while non-µ-opioid receptors mediate SO basal pressures. For unknown reasons, these effects appear to be accentuated in the absence of a gallbladder. One reasonable, but unproven hypothesis is that severing the nerve fibers within the cystic duct during cholecystectomy leads to impaired relaxation of the SO, which compounds the absence of the gallbladder as a pressure reservoir to compensate for increased ductal pressures. In the phase II and III clinical trials of eluxadoline, 10 events consistent with SOS occurred in patients treated with eluxadoline; 8 in the 100 mg group (0.8%) and 2 in the 75 mg group (0.2%). 18 All SOS events occurred in patients without a gallbladder. A total of 8 of the 10 patients with SOS presented with abdominal pain and elevated aminotransferases; 7 in the 100 mg group (0.7%) and 1 in the 75 mg group (0.1%). A total of 75% of these cases occurred within 1 week of study drug initiation. The remaining two cases of SOS included one patient in the 100 mg eluxadoline group with pancreatitis, and one patient in the 75 mg eluxadoline group, with abdominal pain and mild lipase elevation not consistent with pancreatitis.
There were a total of five events meeting the Atlanta criteria for pancreatitis in the phase III trials of eluxadoline that were not consistent with SOS. 18 Three of the five events were associated with heavy alcohol use; two in the 100 mg eluxadoline group and one in the 75 mg eluxadoline group. Of the remaining two pancreatitis events, one was associated with biliary sludge (75 mg) which occurred after 26 weeks of treatment and one occurred in a patient who discontinued eluxadoline 2 weeks prior to the event (100 mg). Both events were deemed unlikely to be study-drug related. All reported pancreatitis events were mild and resolved after discontinuation of the study drug. Given the possible association of eluxadoline with pancreatitis, particularly in those with heavy alcohol intake, it is contraindicated in patients with a history of pancreatitis or active consumption of more than three alcoholic beverages per day.
Based on the safety observations described above, the FDA approved eluxadoline at the lower dose of 75 mg BID in patients without a gallbladder, with recommendations that these patients be monitored closely for abdominal pain, nausea, vomiting and elevations in hepatic or pancreatic enzymes. In Europe and Canada, the absence of a gallbladder is an absolute contraindication for eluxadoline. In the US, since the approval of eluxadoline in May 2015 through February 2017, 120 serious cases of pancreatitis or death were reported via the FDA Adverse Event Reporting System. 20 During this period, approximately 79,600 patients were treated with eluxadoline. Among the 68 patients with reported gallbladder status, 56 did not have a gallbladder. Two deaths occurred in patients with pancreatitis, and a third death occurred in a patient with elevated liver and pancreatic enzymes. All three patients had undergone cholecystectomy. Both patients with pancreatitis who died had additional risk factors for severe pancreatitis; one had a BMI of 38.5 kg/m2 and the other had a history of hereditary pancreatitis and was taking fentanyl and pancreatic enzymes, indicating inappropriate prescribing of eluxadoline in this patient. The third patient suffered a cardiac arrest and had a history of hypertension. In light of this emerging data, a change in FDA labeling, consistent with EMA and Canadian labeling, contraindicating the use of eluxadoline in patients without a gallbladder was recently issued. 20
Abuse potential
Due to the activity of eluxadoline on opioid receptors located on mucosal, myenteric plexus, and smooth muscle cells of the GI tract, it is classified as a schedule 4 medication in the US by the Drug Enforcement Agency. A schedule 4 medication is defined by three criteria: (1) the drug or other substance has a low potential for abuse relative to the drugs or other substances in schedule III; (2) the drug or other substance has a currently accepted medical use in treatment in the US; (3) abuse of the drug or other substance may lead to limited physical dependence or psychological dependence relative to the drugs or other substances in schedule III. In this respect, eluxadoline is similar to other commonly used opioid-receptor agents such as diphenoxylate/atropine and tramadol.
Eluxadoline has an oral bioavailability of less than 1%, likely secondary to minimal absorption and extensive hepatic first-pass metabolism. 14 Approximately 81% is protein bound in systemic circulation and the mean plasma elimination half-life is variable, ranging from 3.7 to 6 hours. Its primary mode of elimination is via the feces as either unabsorbed, unchanged, active drug or via the biliary system, with the kidneys playing only a minor role in drug excretion. Considering the pharmacokinetic profile of eluxadoline and its lack of central pharmacodynamic effects, a low abuse potential would be expected. The abuse potential of either orally or intranasally administered eluxadoline was assessed in nondependent, recreational opioid users in two randomized, double-blinded, placebo- and active-controlled studies. 21 Participants received oral eluxadoline 100, 300 and 1000 mg; oxycodone immediate release 30 and 60 mg; and placebo (oral study) or intranasal eluxadoline 100 and 200 mg; oxycodone immediate release 15 and 30 mg; and placebo (intranasal study). The primary endpoint in both studies was the Drug Liking Visual Analog Scale Maximum Effect score (Drug Liking VAS Emax). Pupillary constriction was also assessed as a measure of central opioid effects. In the oral study, only minimal differences in the Drug Liking VAS Emax were seen following administration of eluxadoline and placebo, with significant differences only observed in the supratherapeutic doses of 300 and 1000 mg. Median Drug Liking VAS Emax scores for oral oxycodone were significantly greater compared with either eluxadoline or placebo. In the intranasal study, median Drug Liking VAS Emax scores for both doses of eluxadoline were not significantly different compared with placebo. This held true despite peak blood concentrations being five- to eightfold higher with intranasal eluxadoline compared with oral eluxadoline. Median Drug Liking VAS Emax scores were significantly higher for both doses of oxycodone compared with both doses of eluxadoline. The mean pupil diameter remained consistent for all doses of oral eluxadoline and was similar to that observed with placebo. Following intranasally administered eluxadoline, the mean pupil diameter decreased for both doses with a significant median difference compared with placebo, however, the median difference in pupil diameter was significantly greater with oxycodone compared with eluxadoline. In addition, euphoric mood was significantly more common with both oral and intranasal oxycodone compared with oral or intranasal eluxadoline.
AEs potentially related to abuse and signs of potential opioid withdrawal were assessed in a pooled analysis of the eluxadoline phase II and both phase III clinical trials by Fant et al. 22 There was no significant difference in the overall incidence of AEs potentially related to abuse between those receiving eluxadoline, both 75 and 100 mg doses, and those receiving placebo (2.7% versus 4.3% versus 2.8%, respectively). Anxiety and somnolence, occurring in <2% of patients in each treatment arm, were the most common AEs potentially related to abuse. Euphoric mood was reported by only two patients, both treated with 100 mg, and resolved without discontinuation of the study drug. The incidence of AEs potentially related to opioid withdrawal were similar across all treatment arms and there was no significant difference among the median overall Subjective Opiate Withdrawal Scale scores for all three groups. In addition, during the 4-week washout period of IBS-3002, no rebound worsening of IBS-D symptoms was reported.
Conclusion
So, what is the place for eluxadoline in the treatment of patients with IBS-D? According to current guidelines, initial therapy of IBS-D should include lifestyle modifications such as dietary manipulation.23,24 Several diets have shown efficacy in patients with IBS, especially IBS-D. These include carbohydrate and gluten-restricted diets (even in patients who do not have celiac disease) and a low-FODMAP diet (limited in fermentable oligo-, di-, and monosaccharides and polyhydric alcohols) that is very restrictive, but has shown benefit in clinical trials. 25 Studies have shown that these diets are optimal when they are initiated and monitored by a trained dietician. 25 Many patients are, however, unable or unwilling to modify their diet, and these diets are certainly not universally effective, even in compliant patients. In addition to the difficulty of complying with some of the IBS diets, insurance companies do not often cover the services of a dietician, and many patients will not pay out of pocket, which is another major limitation to this approach.
In such patients, clinicians typically move to OTC agents, including antidiarrheal medications or probiotics, as well as adjuncts for abdominal discomfort, such as peppermint oil or carminatives. Both the American College of Gastroenterology (ACG) and the American Gastroenterological Association rated the evidence for loperamide in IBS as ‘very low’, with the authors of the ACG guideline stating that there was insufficient evidence to support its use in IBS.24,26 Other IBS guidelines, however, have endorsed the use of loperamide in IBS despite the lack of rigorous clinical trial support, based on the belief that empiric therapy is more likely to deliver benefit than harm in individual patients. 27 A similar pragmatic approach could be taken with probiotics and antispasmodics such as peppermint oil, both of which have demonstrated more convincing results for improving some IBS-D symptoms and are generally well tolerated. Importantly, however, studies have demonstrated that up to 70% of patients who use OTC therapies are dissatisfied with their treatment and most patients presenting to secondary or tertiary care will have tried many of these therapies. 28
From a purely evidence-based clinical practice perspective, the next step in patients with IBS-D who do not respond to lifestyle modifications or OTC therapies should be prescription agents that have demonstrated acceptable efficacy and safety in rigorous clinical trials. There are currently three FDA-approved medications for IBS-D; alosetron (restricted to females with severe IBS-D who have failed conventional therapy), rifaximin, and eluxadoline. While none of these medications can be considered superior to the others due to the absence of direct comparison, all have demonstrated efficacy in the treatment of IBS-D via significant improvements of the primary endpoints compared with placebo (Table 1). Among the prescription therapies with approval for IBS-D, rifaximin and eluxadoline should be used before alosetron. Rifaximin should be used in preference to eluxadoline in patients with contraindications to eluxadoline and is an attractive option, since the treatment duration is only 2 weeks. However, both clinical trial data and clinical experience have demonstrated a high recurrence rate of symptoms among rifaximin responders and many clinicians and patients remain uncomfortable with repeated courses of antibiotics for IBS. Eluxadoline is a logical choice in patients who are unable to obtain rifaximin or who do not respond to an initial course of therapy. Eluxadoline has also been shown to be effective in patients with IBS-D who do not respond to loperamide. 29 Moreover, a recent analysis of the phase III eluxadoline data suggested that only a short course (1 month) of therapy can suggest which patients will have a sustained response with longer-term therapy and also identify sustained nonresponders. 30
Clinical trials of approved prescription IBS-D therapies.

IBS, irritable bowel syndrome; IBS-D, associated with diarrhea; IBS-M, associated with a mixed stool pattern; IBS-C, associated with constipation; FDA, US Food and Drug Administration; BID, twice daily; BSFS, Bristol Stool Form Scale; EMA, European Medicines Agency; TID, three times daily.
Other prescription therapies that are not approved for IBS-D such as tricyclic antidepressants (TCAs) and bile acid sequestrants are also frequently used, but neither class of therapy has been as extensively evaluated as the three approved medications discussed above. Tricyclic antidepressants have been studied in multiple clinical trials and aggregate analyses of this class support their use in IBS; however, they have a narrow therapeutic profile (number needed to harm = 9) 24 and patients are often reluctant to take these medications, even when educated regarding their proposed mechanism of action of modulating enteric nervous system signaling. Bile acid diarrhea has been implicated as a common etiology of IBS-D symptoms, 31 but current level I clinical trial data supporting the use of bile acid sequestrants for IBS-D is lacking.
Ultimately, IBS-D is a heterogeneous disorder and treatment decisions should be individualized on a case-by-case basis, taking into consideration the patient’s symptoms, symptom severity, disease characteristics, coexisting illnesses, and medication cost and safety profile. 14 Given the current efficacy and safety data for rifaximin and eluxadoline in both men and women, it is reasonable to consider both medications as first-line prescription options for IBS-D after lifestyle, dietary modifications and OTC therapies have failed. When prescription therapies are used, emphasis should be placed on appropriate patient selection to optimize clinical efficacy.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
BDC has served as a consultant and speaker for Allergan, PLC, Prometheus Laboratories, and Salix Pharmaceuticals.