
Abstract
Lysosomal acid lipase deficiency (LALD) is a lysosomal storage disorder (LSD) characterized either by infantile onset with fulminant clinical course and very poor prognosis or childhood/adult-onset disease with an attenuated phenotype. The disorder is often misdiagnosed or remains undiagnosed in children and adults due to a rather unspecific clinical presentation with dyslipidemia and steatohepatitis. Until recently, no good treatment options were available for LALD. Despite supportive and symptomatic therapies, death occurred before 1 year of age in patients with infantile-onset disease and patients with childhood/adult-onset LALD suffered from significant complications, such as liver cirrhosis, requiring liver transplantation and early-onset cardiovascular disease.
With the recent approval of sebelipase alfa for clinical use in infantile- as well as childhood/adult-onset LALD, a new treatment era for this disorder has begun. Sebelipase alfa is a recombinant human lysosomal acid lipase (LAL), which is administered via the intravenous route. Clinical trials have shown significant improvement of disease parameters such as liver transaminases, hepatomegaly, and dyslipidemia in childhood/adult-onset LALD patients. Treatment of infants with the severe infantile-onset form of the disease has led to improved survival beyond the age of 1 year, and also showed improvement of hepatic and gastrointestinal symptoms, as well as growth. Overall, sebelipase alfa has a favorable safety profile and promises to be a good long-term treatment option for patients with LALD, with significant reduction of disease burden and increased life expectancy.
Keywords
Introduction
Lysosomal acid lipase deficiency (LALD) is a rare autosomal recessive lysosomal storage disorder (LSD) caused by absent or deficient activity of the enzyme lysosomal acid lipase (LAL). This enzyme plays an important role in the degradation of triglycerides and cholesteryl esters. Decreased LAL enzyme activity leads to pathological accumulation of lipids in macrophages of various tissues throughout the body. 1 Age of onset and phenotypic spectrum are variable and range from an infantile-onset form (Wolman disease) with severe clinical course and death before 1 year of age to childhood/adult-onset disease with milder symptoms, historically also known as cholesteryl ester storage disease. 2 , 3 Both disease entities are part of a continuum and are caused by allelic mutations in the LIPA gene, which encodes the LAL enzyme. 4
While the diagnosis in infantile-onset LALD is usually established early on due to the more uniform and severe clinical course, accurate diagnosis is often missed or delayed in individuals with childhood/adult-onset disease given the less specific and milder clinical presentation. 5
Historically, treatment options for LALD were limited and based on symptom control. Prognosis for patients with infantile-onset LALD was dismal and individuals with childhood/adult-onset LALD were at significant risk for disease complications such as liver cirrhosis requiring liver transplantation at a young age. Additionally, individuals with childhood/adult-onset LALD frequently present with significant dyslipidemia, predisposing them to accelerated atherosclerosis and increasing their risk for cardiovascular disease.5–9 With the recent approval of sebelipase alfa (KanumaTM, Alexion Pharmaceuticals, Cheshire, CT, USA), a recombinant human LAL, a new treatment option for patients with LALD has become available. Clinical trials of this enzyme replacement therapy (ERT) have shown promising results, with significant improvement of symptoms and increased life expectancy in affected individuals.
Clinical presentation
Infantile-onset lysosomal acid lipase deficiency (Wolman disease)
Symptoms of the infantile-onset form of LALD occur in the neonatal period, and newborns frequently present with projectile and persistent vomiting in the first week of life. 3 , 10 , 11 Other common findings are abdominal distention, hepatosplenomegaly, diarrhea, and failure to thrive. Serum transaminases are typically elevated and progressive liver disease with increased bilirubin concentration, coagulopathy, and hypoalbuminemia develops within the first few months of life. Adrenal enlargement with calcification (in 60–70% of cases) visible on abdominal X-ray films is a pathognomonic finding in infantile-onset LALD. 3 , 11 Prognosis is poor, with most infants dying before 1 year of age from malnutrition, liver failure, and adrenal insufficiency. Hematopoietic stem cell transplantation (HSCT) has been attempted, but resulted in early death in most cases.12–14
Childhood/adult-onset lysosomal acid lipase deficiency
The childhood/adult-onset form of LALD was first described in 1963 and is likely underdiagnosed due to its often unspecific and variable clinical presentation. 2 Age of onset ranges from early childhood to adulthood with the majority of affected individuals presenting between 3 and 12 years of age. Diagnostic investigation in children and adults with LALD is often initiated due to significant dyslipidemia at a young age, complaints of abdominal fullness caused by hepatomegaly, or incidentally detected elevated serum transaminases. 5 , 6
A significant delay between occurrence of first symptoms and establishment of the correct diagnosis is common. 5 , 15 Around 30% of children and adults with LALD have been described to develop gastrointestinal symptoms with nausea, vomiting, abdominal pain, and gallbladder dysfunction. 16 However, severe malnutrition and wasting are not usually features of the childhood/adult-onset form of LALD.
The most common disease manifestations of childhood/adult-onset LALD are liver disease and dyslipidemia. Hepatomegaly has been described in 88–99% of cases and splenomegaly has been reported in around 74–79%. 5 , 6 Serum transaminases are frequently elevated, although fluctuation with intermittent normalization of levels has been observed. Hepatic steatosis is common and affected individuals are at a significant risk of developing fibrosis and cirrhosis. Several individuals have been reported who required liver transplantation due to rapid progression to liver failure. 5
Dyslipidemia with patterns consistent with type IIb hyperlipoproteinemia or mixed hyperlipidemia is commonly found in individuals with childhood/adult-onset LALD. Patients typically present with elevated low-density lipoprotein cholesterol (LDL-C) and low high-density lipoprotein cholesterol (HDL-C) levels. Total cholesterol and triglycerides can also be increased. While LDL-C levels often respond to treatment with statins or other lipid-lowering medications, some LALD patients may require higher medication doses or combination of different drugs to obtain a satisfactory response.17–22 In addition, no beneficial effect of statin therapy on hepatic steatosis or fibrosis has been observed. 1 , 6 , 23 , 24 Life expectancy may be decreased due to liver cirrhosis necessitating liver transplantation at a young age. While chronic hyperlipidemia predisposes affected individuals to early-onset atherosclerosis with increased risk for cardiovascular complications, liver disease tends to be the major cause for morbidity and mortality in most of the symptomatic patients with childhood/adult-onset LALD. 5 , 6
Individuals with less severe forms of LALD might have only mild hepatomegaly, intermittent elevation of transaminases, and undetected dyslipidemia. In addition, affected patients with isolated dyslipidemia and absence of hepatomegaly have also been described. 8 , 9 This variable presentation of LALD and significant phenotypic overlap with common disorders such as nonalcoholic fatty liver disease, nonalcoholic steatohepatitis and different forms of dyslipidemia explain why many affected individuals likely remain undiagnosed or are misdiagnosed for many years.
Pathophysiology and prevalence
LALD is an autosomal recessive disorder caused by homozygous or compound heterozygous mutations in the LIPA gene, which encodes the LAL enzyme. LAL plays an important role in the cellular lipid metabolism by degrading cholesteryl esters and triglycerides and thereby providing the cell with free cholesterol and free fatty acids. 25 These intracellular lipids interact with sterol regulatory element-binding proteins (SREBPs) which are transcription factors that modulate expression of genes involved in the lipid metabolism. 26 A deficiency of the LAL enzyme leads to reduced hydrolysis of cholesteryl esters and triglycerides, which subsequently accumulate in the lysosomes. This lysosomal trapping causes a lack of intracellular free cholesterol, which has several consequences for the lipid metabolism: reduced SREBP-mediated feedback inhibition of the enzyme 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase results in increased cholesterol production. Upregulation of LDL-receptor expression occurs, leading to increased cellular uptake of LDL-C. Furthermore, apolipoprotein B, as well as very-low-density lipoprotein cholesterol (VLDL-C) synthesis are markedly increased. 27 This metabolic dysregulation ultimately results in elevated total cholesterol, triglycerides, and LDL-C levels. 25 ,28–30 Low HDL-C concentration is commonly observed in LALD patients and is thought to be the result of impaired upregulation of the ATP-binding cassette transporter A1 (ABCA1) gene in response to intracellular cholesterol. The membrane lipid transporter ABCA1 promotes phospholipid and cholesterol efflux to apolipoprotein A1, which is the rate-limiting step for HDL-C particle formation. In LALD, cholesteryl esters are trapped in the lysosomes and intracellular free cholesterol is reduced. This results in decreased ABCA1 expression and ultimately leads to insufficient availability of extracellular apolipoprotein A1 needed for HDL-C formation.31–33
The accumulation of cholesteryl esters and triglycerides in hepatocytes leads to hepatomegaly and hepatic steatosis. Liver biopsy in patients with childhood/adult-onset LALD typically shows findings consistent with microvesicular steatosis. In addition, foamy macrophages and signs of fibrosis or, in advanced disease, cirrhosis, can be seen on light microscopy. 34
LALD is a pan-ethnic disease, although prevalence among the European White population seems to be higher than in other ethnic groups due to a higher carrier frequency (~1 in 200 individuals) of a recurrent splice-junction mutation E8SJM (c. 894G > A) in exon 8. 35 , 36 This particular mutation is associated with the childhood/adult-onset form of LALD and has never been observed in infantile-onset disease, which is likely explained by higher residual enzyme activity of this allele. Overall, more than 100 pathogenic LIPA mutations have been reported so far. The majority of mutations associated with the infantile-onset form of LALD have been shown to produce LAL enzyme with little or no residual enzymatic activity. 1 , 4 , 6 ,37–40
Diagnosis
The easiest way of establishing the diagnosis of LALD is by measuring the enzymatic activity of LAL in leukocytes. This can be done either on a peripheral blood sample or on a dried blood spot. 41 Confirmation of the diagnosis can be pursued by molecular analysis of the LIPA gene for either homozygous or compound heterozygous pathogenic mutations. While a clear genotype–phenotype correlation cannot be established, individuals with infantile-onset LALD tend to show absent or severely diminished residual enzyme activity. 4
Clinical management
Like many other LSDs, LALD is a multisystemic disorder and should therefore be managed by an interdisciplinary care team. Historically, very limited treatment options for LALD were available, especially for the infantile-onset form of the disease.
Dietary management and supportive care
In infantile-onset disease, nutritional support is crucial and parenteral nutrition is often necessary due to severe malabsorption. However, due to significant comorbidities, malnutrition often cannot be prevented despite early initiation of dietary modifications such as low-fat formula and supplementation with medium-chain triglycerides. 42 , 43
Lipid-lowering medication
In patients with childhood/adult-onset LALD, correction of high LDL-C and low HDL-C levels is often attempted using HMG-CoA reductase inhibitors (statins). While dyslipidemia often improves, therapeutic response is frequently insufficient, requiring high doses or combinations of statins with other lipid-lowering medication such as ezetimibe or cholestyramine.17–22, 24 ,44–46 In addition, statin therapy has not uniformly been shown to have a beneficial effect on hepatic steatosis or serum transaminase concentrations, which often remain elevated despite treatment. 1 , 6 , 23 , 24 In fact, progression of liver disease under statin treatment has been speculated due to upregulation of LDL receptor expression in hepatocytes and subsequently, increased storage of cholesteryl esters. 7
Liver transplantation
Liver transplantation has been reported in several individuals with childhood/adult-onset LALD, most of whom were pediatric patients.47–49 Long-term follow-up information for these patients is limited: while several cases have been reported to be doing well between 10 months and 3 years after transplant, one patient had transplant rejection and developed congestive heart failure. Another individual was found to have significant systemic manifestations of LALD with severe lipid accumulation in the renal vascular system and end stage renal disease 7 years after liver transplantation. 50 , 51
Hematopoietic stem cell transplantation
HSCT has been attempted in several individuals with infantile-onset LALD, and while this is a potentially curative approach, outcomes have generally been poor, with significant comorbidities and a mortality rate of >50%. 14 , 52 , 53 So far, only three children with infantile-LALD have been described in whom HSCT was successfully performed early in life and who were doing reasonably well at 4 and 11 years after transplantation. 13 , 54
Enzyme replacement therapy with sebelipase alfa
General concept
One treatment approach for LSDs is ERT, in which the missing or malfunctioning enzyme is replaced by recombinant enzyme administered usually via the intravenous route. The list of LSDs treatable with ERT is steadily growing as research advances and although the various disorders and therefore enzymatic products are very distinct, they share a common underlying principle. After administration, the recombinant enzyme needs to be taken up by the specific target cells and then directed to the lysosomes, where it can hydrolyze the disease-specific substrate. In order for this process to happen, the recombinant enzyme needs to contain a mannose-6-phosphate moiety that is recognized by mannose-6-phosphate receptors on the surface of the cells targeting the enzyme to the lysosomes. 55
Sebelipase alfa
Sebelipase alfa is a recombinant human LAL that is produced in egg white from transgenic Gallus gallus domesticus hens. The attached mannose-6-phosphate residue facilitates its uptake into the lysosomes of hepatocytes and macrophages where it can subsequently hydrolyze cholesteryl esters and triglycerides. Preclinical studies were performed in a LAL-deficient rat model 56 and showed 100% survival of treated versus untreated animals, as well as a significant improvement of hepatic lipid storage. 57 , 58
Clinical trials in humans subsequently revealed good tolerability of sebelipase alfa and significant improvement of LALD-related symptoms, as well as improved survival in infantile-onset LALD.59–63 The recombinant enzyme was approved for treatment of infantile- as well as childhood/adult-onset LALD by the European Medicines Agency (EMA) in September 2015 and by the US Food and Drug Administration (FDA) in December 2015. Treatment with sebelipase alfa can be initiated as soon as the diagnosis of LALD is established and the recombinant enzyme is administered via the intravenous route. The recommended dosing is 1 mg/kg weekly for infantile-onset LALD and 1 mg/kg every 2 weeks in individuals with childhood/adult-onset LALD. 64 , 65
Sebelipase alfa: clinical trials
To assess safety profile as well as efficacy of treatment with sebelipase alfa, separate clinical trials were conducted or are still ongoing for infantile- and childhood/adult-onset LALD (Table 1).
Outline of the different clinical trials with sebelipase alfa conducted in individuals with infantile- and childhood/adult-onset lysosomal acid lipase deficiency.
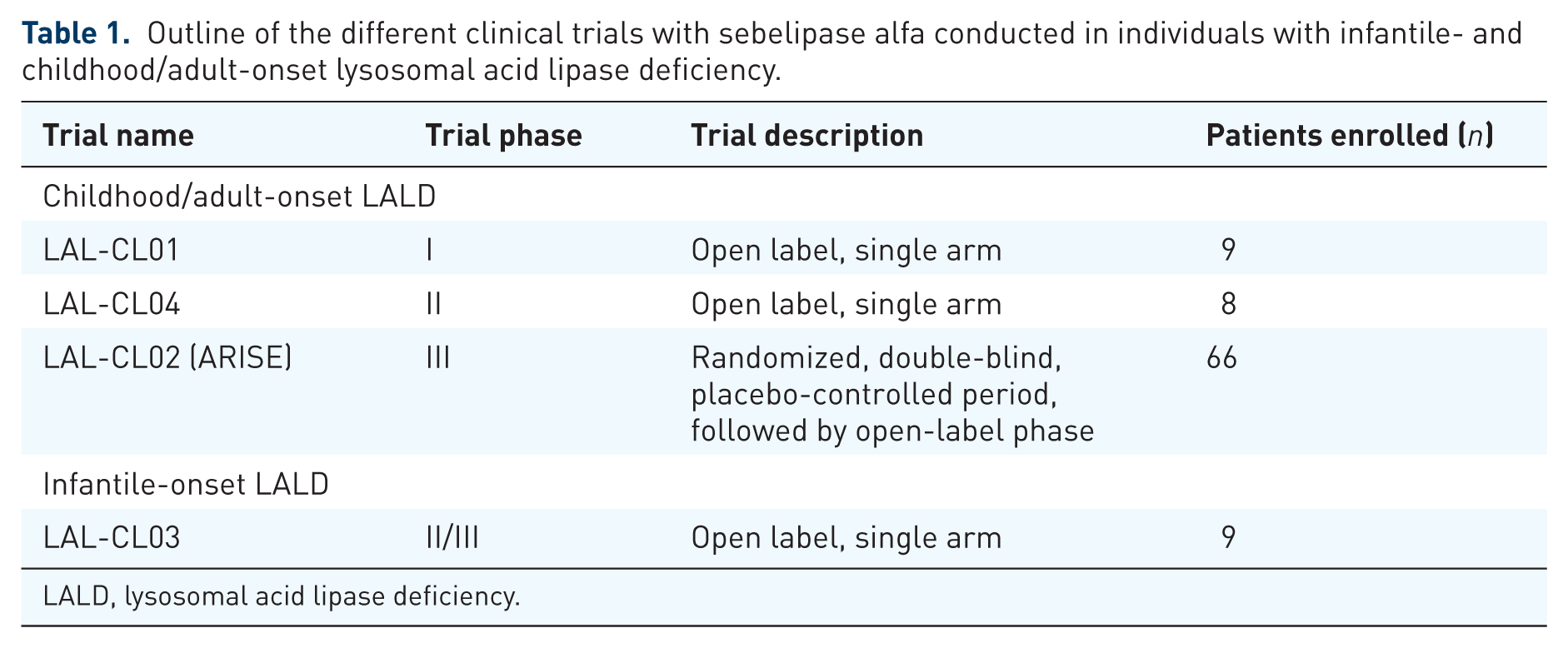
LALD, lysosomal acid lipase deficiency.
Childhood/adult-onset lysosomal acid lipase deficiency
The first use of sebelipase alfa in humans was investigated in an open-label phase I clinical trial (LAL-CL01) [ClinicalTrials.gov identifier: NCT01307098]. Nine adult individuals with clinical manifestations of LALD received weekly infusions of sebelipase alfa at different doses (0.35, 1, or 3 mg/kg) over 4 weeks. Outcome measures included safety assessment as well as pharmacodynamic studies. Results revealed a favorable safety profile as well as a statistically significant reduction of liver transaminases in all study subjects. 59
Of the nine individuals in LAL-CL01, eight were enrolled in the extension (phase II) clinical trial LAL-CL04 [ClinicalTrials.gov identifier: NCT01488097], which was also an open-label trial. After a washout period of 9–28 weeks, the patients again received weekly sebelipase alfa infusions at the same doses of 0.35, 1, or 3 mg/kg for 4 weeks. After this initial period, they continued to be treated with either 1 or 3 mg/kg recombinant enzyme at biweekly infusion intervals for a total of 52 weeks. During the treatment-free interval between LAL-CL01 and LAL-CL04, patients’ transaminases had reached pretreatment levels. After re-initiation of sebelipase alfa treatment in LAL-CL04, liver enzymes decreased at a similar rate to what had been observed in the phase I trial, and remained stable over the prolonged treatment interval of 52 weeks even after infusion frequency was switched to biweekly. At that time, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) concentrations showed a mean change from baseline of –58% and –40%, respectively. In addition, lipid parameters were assessed and while total cholesterol and LDL-C concentrations had initially increased after 4 weeks of treatment, mean concentrations for both had changed by –39% and –60%, respectively when compared with baseline. HDL-C concentration had changed +29% from baseline. It is speculated that the temporary increase in total cholesterol and LDL-C shortly after ERT initiation was caused by mobilization of accumulated lipids from liver, spleen as well as other tissues. As in the phase I trial, none of the patients experienced clinically concerning adverse events and no antibodies against the infusion drug could be detected. 61
The phase III clinical trial (LAL-CL02 or ARISE) [ClinicalTrials.gov identifier: NCT01757184] was a multicenter, randomized, double-blind, placebo-controlled study. A total of 66 patients with childhood/adult-onset LALD were randomized to placebo or sebelipase alfa infusion at 1 mg/kg every other week. The placebo-controlled part of the trial was 20 weeks, followed by an open-label period during which all patients received treatment. The primary endpoint was normalization of ALT concentration and secondary endpoints included further disease-related outcomes, safety, and side-effect profile.
After completion of the placebo-controlled part, 31% of patients treated with sebelipase alfa had reached the primary endpoint as compared with 7% in the placebo group (p = 0.03). Results for AST were similar. Consistent with the improvement in liver function, a significant reduction of hepatic fat content compared with baseline was observed (–32% in sebelipase alfa versus –4.2% in placebo group, p = 0.001). This was determined using multi-echo gradient-echo MRI. For assessment of severity of microvesicular steatosis, liver biopsies at baseline and at 20 weeks were obtained in 16 patients in the sebelipase alfa group and in 10 patients who received placebo. While a reduction in steatosis was more frequently observed in the treatment group than in the placebo group, this finding did not reach statistical significance.
A positive effect of sebelipase alfa treatment on the lipid profile was also reported. While LDL-C improved by –28.4% from baseline in the treatment group, a change of only –6.2% from baseline was seen in the placebo group (p < 0.001). Similar findings were present for HDL-C, which changed by +19.6% from baseline in patients treated with sebelipase alfa compared with –0.3% in the placebo group (p < 0.001).
During the open-label period, after switching from placebo to sebelipase alfa treatment, the pattern of changes in ALT, LDL-C, and HDL-C was very similar to what was observed in the initial sebelipase alfa group. In addition, a further reduction in LDL-C was achieved in the initial treatment group with continued sebelipase alfa infusions.
Overall, sebelipase alfa was well tolerated and only one atypical infusion-related event was reported. Out of the 35 patients in the sebelipase alfa group, five developed antibodies against the infusion drug, but titers were low and nonsustained. They were not found to have an effect on safety or efficacy of sebelipase alfa. 60
Infantile-onset lysosomal acid lipase deficiency
A phase II/III clinical trial (LAL-CL03) [ClinicalTrials.gov identifier: NCT01371825] investigating the use of sebelipase alfa in patients with infantile-onset LALD is an open-label trial conducted in Europe, the USA and the Middle East. All patients receive treatment with sebelipase alfa. The primary endpoint is survival at age 12 months and results are compared with data from a historical cohort. Secondary endpoints include safety, growth and liver function, as well as hematological parameters.
By the time of the last report in July 2015, five of the initially nine enrolled patients were still on ERT with sebelipase alfa. Three children had died due to causes unrelated to the infusion drug; the reason for discontinuation of treatment in the fourth child was not reported. All patients started weekly sebelipase alfa infusions at 0.2 or 0.35 mg/kg and the dose was then escalated to 1 or 3 mg/kg per week. The primary endpoint was met in all five patients and their ages ranged between 2 years 5 months and 4 years 7 months at the time the results were published. An improvement was observed of ALT and AST (median percent changes of –45.59% and –39.36% from baseline, respectively) and hepatosplenomegaly as well as gastrointestinal symptoms had improved. In addition, the children’s median weight percentile had increased from 3.59% at baseline to 35.09%. Out of five patients, four had normal scores when evaluating their development using the Denver II screening test.
ERT was reported to be overall well tolerated, with only one patient experiencing a treatment-related serious adverse event consisting of fever, chills, pallor, and tachycardia. Antibodies against the infusion drug were detected in four out of seven patients tested and they were found to be neutralizing in two of them. However, neither the infusion-related reaction nor the development of antibodies led to discontinuation of ERT. 62 , 63
Discussion
LALD is an LSD with variable age of onset and varying degrees of phenotypic severity. It is a rare disorder but owing to the unspecific clinical presentation of the childhood/adult-onset type, LALD is very likely underdiagnosed and its prevalence is expected to be higher than previously reported. 35 , 36 Until the recent EMA and FDA approval of sebelipase alfa, treatment was mainly symptomatic and prognosis for the infantile-onset form of LALD was dismal, with death usually before age 1. Patients with childhood/adult-onset LALD may have decreased life expectancy due to significant liver disease, often requiring liver transplantation at a young age. In addition, accelerated atherosclerosis and an increased risk for early-onset cardiovascular disease are a concern in affected individuals due to unfavorable lipid profiles. 5 , 6 , 8 , 9 While so far, no long-term outcome data for treatment with sebelipase alfa is available, clinical trials showed significant reduction of disease burden in both, infants and individuals with childhood/adult-onset disease. This led to significant improvement of survival in the infantile-onset group and is expected to also increase life span and quality of life in patients with childhood/adult-onset LALD. However, longer-term follow up is needed to establish the full extent to which children with infantile-onset LALD benefit from treatment and whether complications such as liver cirrhosis and cardiovascular events can truly be prevented over time in patients with childhood/adult-onset disease.
It should also be noted that individuals with advanced liver disease were not included in the clinical trials of childhood/adult-onset LALD. It is therefore undetermined and remains to be evaluated whether treatment with sebelipase alfa can halt or reverse symptoms of advanced liver disease.
While animal studies did not show a harmful effect on embryogenesis or fetal development, no experience during pregnancy in humans is available so far and no recommendations regarding sebelipase alfa treatment and dosing can therefore be provided at this time. 65
In conclusion, sebelipase alfa has been shown as an efficient and safe treatment for infantile- as well as childhood/adult-onset LALD but long-term outcomes remain to be determined. Infantile-onset LALD is usually more easily diagnosable due to the severity of the clinical presentation, and treatment with sebelipase alfa should be initiated as soon as the diagnosis is established by enzymatic screening. Childhood/adult-onset LALD, however, often presents with unspecific clinical features and is therefore frequently undiagnosed or misdiagnosed. With the availability of sebelipase alfa, establishing the correct diagnosis as early as possible is crucial, and broad enzymatic screening for LALD in children and adults with hepatic disease and/or dyslipidemia is recommended. However, given the rarity of this disorder, disease awareness among healthcare providers is low and LALD is often not part of the differential diagnosis. Education, as well as access to enzymatic screening will therefore prove essential to ensure that patients with childhood/adult-onset LALD are diagnosed and started on treatment in a timely fashion.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Dr Erwin has served as an advisory board member for Alexion Pharmaceuticals.