
Abstract
Background:
Dual delayed-release dexlansoprazole is approved for use in adults as a 30 mg orally disintegrating tablet (ODT) or as 30 mg and 60 mg capsules. The pharmacokinetics, pharmacodynamics, and safety profile of two dexlansoprazole 30 mg ODTs were compared with one dexlansoprazole 60 mg capsule in this randomized, phase I, open-label, single-center, multiple-dose, two-period crossover study.
Methods:
Participants were randomized in one of two treatment sequences, each comprised two 5-day treatment periods during which two dexlansoprazole 30 mg ODTs or one 60 mg capsule was administered once daily. Pharmacokinetic parameters and the mean intragastric pH profile for the 24-hour period after dosing on days 1 and 5 were described. Adverse events were monitored during study duration and followed up with a phone call 5–10 days after the last dose of study drug.
Results:
On day 1, peak observed plasma concentration (Cmax) values were similar between two 30 mg ODTs (1047 ng/ml) and one 60 mg capsule (1164 ng/ml). Systemic exposure, measured by the area under the plasma concentration–time curve (AUC), was approximately 25% lower after ODT administration. On day 5, mean pH after daily doses of two 30 mg ODT or one 60 mg capsule was 4.33 and 4.36, respectively; both regimens maintained intragastric pH above 4.0 for 60% of the 24-hour period. Headache was the most commonly reported adverse event (observed in 19.2% of participants); no adverse events leading to study withdrawal occurred.
Conclusions:
While systemic exposure (AUC) was 25% lower with ODT, peak concentrations (Cmax) after administration of two dexlansoprazole 30 mg ODTs and one 60 mg capsule were similar. The 24-hour intragastric pH control after administration of two dexlansoprazole 30 mg ODTs was equivalent to one dexlansoprazole 60 mg capsule. Both ODT and capsule were well tolerated.
Keywords
Introduction
Gastroesophageal reflux disease (GERD) is a chronic digestive disorder caused by inadequate closure of the lower esophageal sphincter and results in acidic stomach contents inappropriately entering the esophagus, leading to heartburn and other troublesome symptoms [Zhao and Ecinosa, 2008; Saraf et al. 2012]. GERD affects up to 20% of adults in the US [Dent et al. 2005]; when untreated, chronic acid regurgitation can damage the lining of the esophagus and some patients may develop erosions of the esophageal mucosa [erosive esophagitis (EE)] [Saraf et al. 2012; Katz et al. 2013]. Though changes in diet are recommended for GERD management, many must resort to pharmacologic treatment with acid-suppressing agents, such as antacids, histamine-2 receptor antagonists, or proton pump inhibitors (PPIs) for symptom relief and esophageal mucosal healing [Katz et al. 2013].
PPIs are the treatment of choice for healing of EE and relief of GERD symptoms because of their inhibitory action on the hydrogen/potassium adenosine triphosphatase enzyme (proton pump), the final site of acid secretion in the parietal cell [Katz et al. 2013; Shin and Kim, 2013]. Unlike other drugs in this class, the PPI dexlansoprazole is formulated as a capsule with a dual delayed-release mechanism [Vakily et al. 2009; Takeda Pharmaceuticals America, Inc., 2016]. According to the dual delayed-release mechanism, dexlansoprazole is first released 1–2 hours after ingestion, followed by a second release within 4–5 hours. This dual delayed-release formulation extends the duration of drug exposure and maintains pharmacologically active levels of drug over a longer period of time, resulting in prolonged elevation of intragastric pH [Vakily et al. 2009]. The prolonged acid suppression addresses potential breakthrough heartburn that may occur at night [Fass et al. 2009; Takeda Pharmaceuticals America, Inc., 2016].
The pharmacokinetic, pharmacodynamic, efficacy, and safety profiles of dexlansoprazole capsule after administration of doses of 30 and 60 mg have been extensively studied in adults in randomized, double-blind, controlled clinical studies [Peura et al. 2009; Wittbrodt et al. 2009]. Patients with symptomatic nonerosive GERD experienced medians of 50.0–54.9% of 24-hour days and 76.9–80.8% of nights without heartburn over 4 weeks with both doses [Fass et al. 2009]. In two separate studies, complete healing of EE was observed in 92% and 93% of patients receiving 8-week treatment with the 60 mg dexlansoprazole capsule [Sharma et al. 2009]. In patients with healed EE, both 30 and 60 mg strengths of the dexlansoprazole capsule resulted in medians of 91–96% of 24-hour days and 96–99% of nights without heartburn over 6 months [Metz et al. 2009]. In a pooled safety study of six controlled trials, patients receiving the dexlansoprazole capsule also had lower rates of treatment-emergent adverse events per 100 patient-months (15.64–18.75%) than those receiving placebo (24.49%) or lansoprazole (21.06%) [Peura et al. 2009].
Most PPI formulations need to be swallowed whole and may be uncomfortable to ingest for patients with difficulty swallowing, otherwise known as dysphagia [Horn and Howden, 2005; Katz et al. 2013; Liu et al. 2014]. A GERD diagnosis and PPI use are both significantly associated with frequent dysphagia [Cho et al. 2015]. Further, in a population-based study, patients with symptoms of heartburn and acid reflux were almost five times more likely to report dysphagia than those without symptoms [odds ratio (OR) 4.7; 95% confidence interval (CI) 2.9, 7.4] [Locke et al. 1997]. Dysphagia is also commonly associated with many neurologic disorders, with prevalence rates of comorbid dysphagia of over 30% each among patients with stroke, dementia, Alzheimer’s dementia, Parkinson’s disease, Huntington’s disease, multiple sclerosis, and amyotrophic lateral sclerosis [Daniels, 2006]. In the general population, dysphagia is reported by about 20% of Americans [Cho et al. 2015].
Patients with difficulty swallowing find orally disintegrating tablets (ODTs) easier to swallow than conventional tablets [Carnaby-Mann and Crary, 2005]. Nearly 75% of patients with difficulty swallowing prefer ODTs, and their administration has been found to result in shorter swallowing duration, less muscular effort, and less fluid assistance requirement than with ingestion of a standard tablet [Carnaby-Mann and Crary, 2005].
Until recently, lansoprazole has been the only PPI available as an ODT. A dual delayed-release ODT formulation of dexlansoprazole developed to disintegrate in the mouth is an attractive option for patients with difficulty swallowing capsules who require acid-suppressive therapy. The dexlansoprazole ODT formulation also showed flexibility in dosing after bioequivalence was demonstrated between standard ODT administration and oral delivery of the disintegrated tablet in water via syringe or nasogastric tube [Kukulka et al. 2014]. The bioequivalence between the dexlansoprazole 30 mg ODT and 30 mg capsule has been demonstrated [Kukulka et al. 2015], and the 30 mg ODT formulation of dexlansoprazole was recently approved in the US for the same indications for which the 30 mg dexlansoprazole capsule is approved, that is, for treating heartburn associated with symptomatic nonerosive GERD for 4 weeks, and for maintenance of healed EE and relief of heartburn for up to 6 months. For the healing of EE, the currently approved dexlansoprazole formulation is the 60 mg capsule, administered once daily for 8 weeks [Takeda Pharmaceuticals America, Inc., 2016]. This study compared the bioavailability, pharmacodynamics, and safety profiles after administration of two dexlansoprazole 30 mg ODTs and one dexlansoprazole 60 mg capsule.
Methods
The phase I, randomized, open-label, single-center, multiple-dose, two-period crossover study was conducted at Celerion in Tempe, AZ, from January to April 2014 [ClinicalTrials.gov identifier: NCT02064907]. Study protocols were developed according to the Food and Drug Administration Guidance for Industry: Bioavailability and Bioequivalence Studies for Orally Administered Drug Products [US Food and Drug Administration, 2003], approved by the Independent Investigational Review Board, and conducted compliant with the Declaration of Helsinki and the International Conference on Harmonisation (ICH) Harmonised Tripartite Guideline for Good Clinical Practice (GCP) [International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, 2001; World Medical Association, 2013].
Study participants
Participants aged 18–55 years with a body mass index between 18 and 30 kg/m2, a weight of at least 50 kg, and in good health, as evidenced by clinical chemistry, hematology, and complete urinalysis results within normal levels were eligible for enrollment into this study. Participants were ineligible if they had any clinically significant comorbidity that could potentially confound study results or impact their ability to participate in the study. Participants were required to use adequate contraception and female participants were required to have a negative serum pregnancy test at screening. Additional exclusion criteria included prior use of dexlansoprazole or lansoprazole or a known hypersensitivity to any component of the dexlansoprazole ODT, dexlansoprazole capsule, or other PPI. The study also prohibited concomitant or prior administration of agents that could potentially affect drug metabolism or clearance, including, but not limited to, products containing caffeine/xanthine, nicotine or grapefruit products. Participants with a history or evidence of alcohol or drug abuse were also excluded from the study. Before initiating any study procedures, written informed consent was obtained from the study participants. If any of the exclusion criteria were violated after randomization, the participant was removed from the study.
Study design
Participants were randomized 1:1 to sequence one or sequence two (Figure 1). Each sequence comprised two treatment periods separated by a washout period of at least 7 days. In sequence one, participants received two dexlansoprazole 30 mg ODTs once daily for 5 days during the first treatment period and a single dose of the dexlansoprazole 60 mg capsule in the second treatment period. In sequence two, study participants received the capsule and ODTs in the reverse order. There was a minimum 7-day washout period between the last dose in the first period and the first dose of the second period. Participants were confined to the clinic during each treatment period.

Schematic of study design. The trial was designed as a 2-period crossover study. Participants were randomized to one of two treatment sequences. Within each treatment sequence, participants received daily doses of two dexlansoprazole 30-mg ODTs and one dexlansoprazole 60-mg capsule during two treatment periods separated by a 7-day washout period. Participants were confined to the clinic from day −1 to day 6 of each treatment period and followed up with a phone call 5 to 10 days after the last dose of study drug.
The two dexlansoprazole 30 mg ODTs were administered as described previously for a single ODT [Kukulka et al. 2015]. Briefly, the first ODT was administered on the tongue without water and participants were instructed to allow the tablet to completely disintegrate before swallowing the granules without chewing. The second ODT was administered immediately after the first ODT was swallowed. Participants were instructed to swallow the dexlansoprazole 60 mg capsule intact with 240 ml of water. Participants could drink water at any time outside of a specified 2-hour window 1 hour before and after treatment administration. Both formulations were administered after an overnight fast of at least 10 hours, followed by a 4-hour refrain from meals after receiving doses on days 1 and 5 and a 1-hour refrain after dosing on days 2, 3, and 4.
Sample collection
For pharmacokinetic analyses, 3-ml blood samples were collected on days 1 and 5 within 30 minutes before dose administration and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 16, and 24 hours after dosing. Dexlansoprazole is a substrate for polymorphic cytochrome P450 (CYP) 2C19, and so, CYP2C19-deficient individuals would be expected to have higher plasma concentrations of dexlansoprazole [Takeda Pharmaceuticals America, Inc., 2016]. Therefore, an additional 6 ml blood sample was collected on day 1 of treatment period one to assess CYP2C19 genotype and metabolizer status in all participants. Blood samples that were not collected at the designated time point were noted on the electronic case report form. A validated liquid chromatography tandem mass spectrometry assay (PPD, Inc., Middleton, WI) with a detection range of 2.00–2000 ng/ml was used to determine the plasma concentrations of dexlansoprazole. Concentrations lower than 2.00 ng/ml were treated as zero for the pharmacokinetic analyses.
For pharmacodynamic analyses, a single-channel antimony probe attached to a data recorder unit (Sandhill Scientific, Inc., Highlands Ranch, CO) was inserted into the stomach to a predetermined length (about 10 cm) past the lower esophageal sphincter. Standard clinical procedures were employed throughout this process, which included allowing participants to drink water and to use topical anesthetics to aid in the placement of the pH probe. The probe was calibrated with standard buffers before each use. On days 1 and 5, the intragastric pH was sampled and recorded every 5 seconds for 24 hours, beginning immediately before the study drug was administered.
Pharmacokinetic parameters
The primary objective of the study was to assess the pharmacokinetics of two dexlansoprazole 30 mg ODTs versus one 60 mg capsule on days 1 and 5. The primary endpoints were the maximum observed plasma concentration (Cmax) of dexlansoprazole and area under the plasma concentration-time curve (AUC), a measurement of systemic drug exposure. AUClast (AUC from time 0 to the time of last quantifiable concentration) was used to measure systemic exposure after a single dose on day 1 and multiple doses on day 5 of each treatment period. The AUC from time 0 to infinity, or AUC ∞ , was calculated on day 1 of each treatment period only, while the AUC during a specified dosing interval, or AUCtau, was used to calculate exposure only on day 5. Additional pharmacokinetic endpoints included the rate of absorption, defined as the time required to reach Cmax (Tmax), the apparent clearance after extravascular administration (CL/F), the terminal elimination half-life (T1/2), and the apparent volume of distribution (Vz/F). All pharmacokinetic parameters were derived from plasma concentrations measured on days 1 and 5 of each treatment period and were estimated using Phoenix WinNonlin Version 6.3 software (Certara, Princeton, NJ).
Pharmacodynamic parameters
Pharmacodynamic profiles of two dexlansoprazole 30 mg ODTs and one 60 mg capsule were investigated as an additional objective of the study. The mean intragastric pH and percentage of time with intragastric pH above 4 during the 24-hour period after dose administration on days 1 and 5 were evaluated.
Safety endpoints
The secondary objective of the study was to evaluate the safety profiles of two dexlansoprazole 30 mg ODTs and one 60 mg capsule. Treatment-emergent adverse events were monitored through physical examination, clinical laboratory testing, vital sign measurement, 12-lead electrocardiogram, and self-report. Adverse events were classified according to the Medical Dictionary for Regulatory Activities’ preferred term (Version 16.1) [Brown et al. 1999] and classified as serious if they were life threatening or resulted in death, hospitalization, or incapacitation, or were deemed an important medical event by investigators. Adverse events were monitored during confinement, and any ongoing or emerging adverse events were followed up with a phone call 5–10 days after the last dose of study drug was administered.
Statistical analysis
All data analyses were generated using SAS (Statistical Analysis System) version 9.2 (SAS Institute, Cary, NC). Descriptive statistics including the mean, standard deviation, percent coefficient of variation (%CV), and median (minimum and maximum) values were generated for individual pharmacokinetic parameters of dexlansoprazole by day. For relevant computations, actual sampling times were used over scheduled sampling times. Dexlansoprazole Cmax and AUC parameters for the two formulations were further characterized as geometric means and as individual ratios on the original scale and the difference on the natural log scale. Participants with available data for both treatment regimens (ODT and capsule) were included in the pharmacokinetic analyses.
In the crossover study design, each participant received both regimens. While genetic variations between individuals, including CYP polymorphisms, can affect bioavailability estimates, the basic bioavailability/bioequivalence study design used here compared the formulations against each other within each individual. Since the bioavailability following administration of each formulation would be affected equally within an individual, a participant’s CYP2C19 genotype would not affect the statistical assessments of equivalence. Therefore, no formal statistical analyses were conducted based on CYP2C19 genotype.
The 24-hour intragastric pH profile after dosing on days 1 and 5 was derived from pH values collected every 5 seconds. The median pH was calculated every 15 minutes to reduce the inherent variability in pH measurements, and the mean pH over the total 24-hour period was derived as the average of the median pH values. The percentage of time with pH above 4 over a time period was calculated as the percentage of total 15-minute medians that had a pH value above 4 for that period.
Both pharmacokinetic (Tmax and natural log-transformed Cmax and AUC) and pharmacodynamic (mean pH and % of time pH > 4) parameters were analyzed using analysis of variance (ANOVA) models on days 1 and 5. Sequence, treatment period, regimen (ODT or capsule), cohort, and the interaction between cohort and period were included in the model as fixed factors and the participant nested within sequence and cohort as a random factor.
Derived from the ANOVA model, point estimates of the relative bioavailability of dexlansoprazole when administered as two 30 mg ODTs were compared with the bioavailability of dexlansoprazole when administered as a single 60 mg capsule. Bioequivalence between the two 30 mg ODTs and one 60 mg capsule was concluded if the corresponding 90% CIs for the pairwise comparisons for Cmax and AUC were confined within the bioequivalence range of 0.80–1.25 [US Food and Drug Administration, 2003]. The effect between single and multiple daily doses of either ODT (2 × 30 mg) or capsule (1 × 60 mg) was evaluated using a paired t-test on Tmax and natural log of Cmax and AUC; no effect was concluded if the 90% CIs for the ratio of Cmax and AUC central values were within the 0.80–1.25 interval. For the percentage of time that pH was above 4, pharmacodynamic equivalence between the ODT and capsule formulations was concluded if the 90% CIs for the difference were within the prespecified range of −12% to 12%. This range was determined with consideration of relevant variance measures in similar studies [Hartmann et al. 1998; Simon et al. 2003; Armstrong et al. 2007].
Results
Study population
A total of 52 healthy adults were enrolled in the study, with 26 randomized to each treatment sequence. Similar baseline characteristics were observed between the two treatment-sequence groups (Table 1). The mean age of participants was 37.6 years, and most participants were White and of Hispanic ethnicity. Determination of the CYP2C19 genotype showed that 37 of the 52 enrolled participants were extensive metabolizers, 14 were ultra-rapid metabolizers, and the remaining participant was a poor metabolizer.
Baseline characteristics.

BMI, body mass index; SD, standard deviation.
In sequence 1, participants received a daily dose of two dexlansoprazole 30 mg ODTs for 5 days followed by a daily dose of one dexlansoprazole 60 mg capsule for 5 days.
In sequence 2, participants received daily doses of one dexlansoprazole 60 mg capsule for 5 days followed by a daily dose of two dexlansoprazole 30 mg ODTs for 5 days.
Age at first dose of study drug.
Pharmacokinetics
The pharmacokinetic parameter estimates, after administration of two dexlansoprazole 30 mg ODTs or one dexlansoprazole 60 mg capsule, were determined on day 1 as well as after 5 daily doses of each regimen on day 5. Drug absorption was faster with the ODT when administered as two 30 mg ODTs than with the 60 mg capsule, and mean dexlansoprazole Cmax values were similar between the two 30 mg ODTs and the 60 mg capsule (Table 2). However, bioequivalence was not established because the overall systemic exposure, as measured by AUC, was approximately 22–25% lower after ODT (2 × 30 mg) administration (Table 3).
Pharmacokinetic parameter estimates after single administration of 60 mg dexlansoprazole.

Note: Because of variability in the terminal phase of the plasma concentration–time curve, the terminal elimination rate constant could not be determined for some subjects, and therefore the pharmacokinetic parameters that use this constant in their calculations (i.e. T1/2, AUC∞, CL/F, and Vz/F) could not be estimated.
AUC∞, area under the plasma concentration–time curve from time 0 to infinity; AUCtau, area under the plasma concentration–time curve during a dosing interval; CL/F, apparent clearance after extravascular administration; Cmax, maximum observed plasma concentration; ODT, orally disintegrating tablet; T1/2, terminal elimination half-life; Tmax, time to reach maximum observed plasma concentration; SD, standard deviation; Vz/F, apparent volume of distribution after extravascular administration.
Median (minimum–maximum) reported for Tmax.
AUC ∞ reported for day 1 and AUCtau reported for day 5.
Statistical comparison of pharmacokinetic parameters after administration of 60 mg dexlansoprazole.

AUC∞, area under the plasma concentration–time curve from time 0 to infinity; AUCtau, area under the plasma concentration–time curve during a dosing interval; AUClast, area under the plasma concentration–time curve from time 0 to the last quantifiable concentration; CI, confidence interval; Cmax, maximum observed plasma concentration; n/a, not applicable; ODT, orally disintegrating tablet.
The effect of repeat administration of either formulation given as two 30 mg ODTs or one 60 mg capsule was evaluated by comparing the Cmax and AUC estimates on days 1 and 5. The corresponding 90% CIs for both Cmax and AUC were within bioequivalence limits (Table 3), indicating that multiple dosing had no effect on dexlansoprazole pharmacokinetics.
Pharmacodynamics
On day 5, similar pH profiles for the 24 hours after daily administration of two 30 mg ODTs or the 60 mg capsule were observed (Figure 2). The mean intragastric pH during the 24-hour period after the fifth daily dose of two 30 mg ODTs or the 60 mg capsule was 4.3 and 4.4, respectively, and both formulations maintained an intragastric pH above 4 for 60% of the 24-hour postdose time interval (Figure 3). Intragastric pH was also maintained above 4 for 61% and 62% of the >6- to 24-hour window, including the overnight hours, after administration of the two 30 mg ODTs and 60 mg capsule, respectively (90% CI for the difference −4.0, 1.8; data on file).

Mean pH profiles for the 24-hour time period postdose on day 5. Dexlansoprazole (60 mg) was administered daily as two 30-mg ODTs (black) or one 60-mg capsule (blue) for 5 days. Median intragastric pH values over 15-minute time intervals for the 24-hour time period postdose were determined from pH readings taken every 5 seconds.
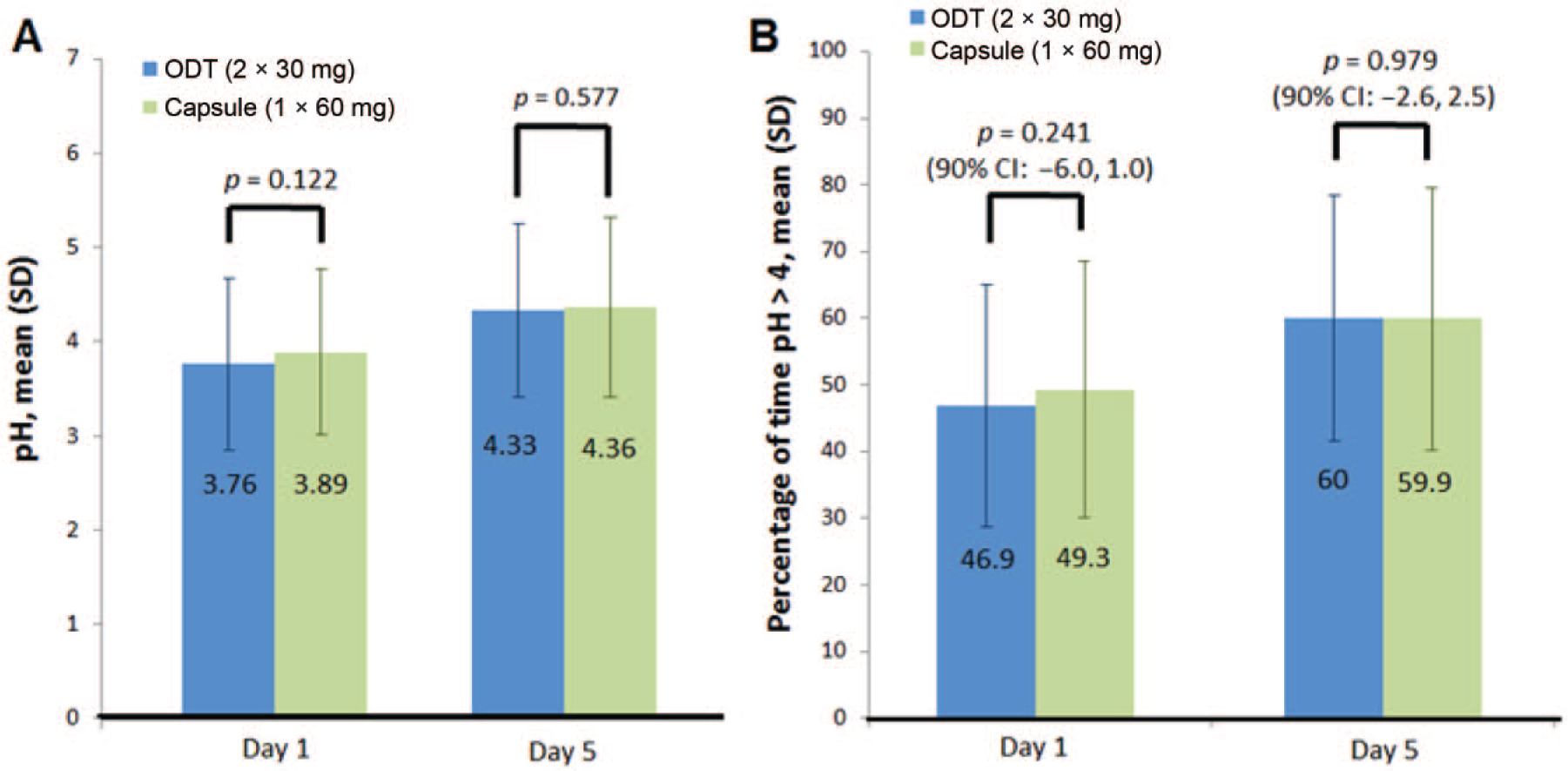
Pharmacodynamic evaluation of 60-mg dexlansoprazole administered daily as two 30-mg ODTs or one 60-mg capsule on days 1 and 5. (A) Mean pH and (B) percentage of time intragastric pH > 4 over 24 hours. Error bars indicate standard deviation.
Similar pH profiles were observed on day 1 after a single dose of two dexlansoprazole 30 mg ODTs or 60 mg capsule. However, while the mean pH and the percentage of time with an intragastric pH above 4 after a single daily dose were comparable between the two formulations, both parameters were lower than those observed on day 5 after repeat dosing (Figure 3).
Summary of adverse events
Adverse events were reported by 33% of participants when receiving a dose of two dexlansoprazole 30 mg ODTs and 35% when receiving the dexlansoprazole 60 mg capsule. Most adverse events (49/52) were classified as mild, and the majority (38/52) were considered unrelated to the study drug. Headache, nausea, and dizziness occurred in five (9.6%), three (5.8%), and two (3.8%) participants, respectively, when receiving the two 30 mg ODTs. Headache, decubitus ulcer, oropharyngeal pain, and flatulence were reported in six (11.5%), three (5.8%), two (3.8%), and two (3.8%) participants when receiving the 60 mg capsule. Adverse events experienced by one (1.9%) participant included oropharyngeal pain and flatulence with the two ODTs and nausea with the capsule. There were no deaths or serious adverse events recorded, and no adverse events led to withdrawal from the study.
Discussion
The relative bioavailability, safety profile, and pharmacodynamics of two dexlansoprazole 30 mg ODTs and a single 60 mg capsule were evaluated in this study. Both formulations employ a dual delayed-release mechanism and contain two types of enteric-coated granules that release drug at different pH values after dosing. The ODT and capsule formulations of 30 mg dexlansoprazole are approved for the treatment of heartburn associated with symptomatic nonerosive GERD for 4 weeks, and maintenance of healed EE and relief of heartburn for up to 6 months. Dexlansoprazole delayed-release capsules are also indicated for the healing of all grades of EE for up to 8 weeks when dosed at 60 mg once daily [Takeda Pharmaceuticals America, Inc., 2016].
Bioequivalence between the 30 mg dexlansoprazole ODT and the 30 mg dexlansoprazole capsule has been demonstrated by similar Cmax (688 and 618 ng/ml for the ODT and capsule, respectively) and AUC values (3048 and 3212 ng·hour/ml for the ODT and capsule, respectively) [Kukulka et al. 2015]. The dexlansoprazole 30 mg ODT and 30 mg capsule also maintained intragastric pH above 4 for 45.8% and 47.3% of a 24-hour period, respectively, indicating equivalent pH control throughout the day [Kukulka et al. 2015].
Patients with difficulty swallowing find ODT formulations much easier to swallow, with one study citing reduced physiologic effort in swallowing with no increase in airway compromise, and 76% of dysphagic patients preferring ODT medication delivery to the conventional tablet [Carnaby-Mann and Crary, 2005]. Inability to swallow can impact medication compliance, which can adversely increase patient morbidity [Carnaby-Mann and Crary, 2005]. Affecting a sizable portion of the US population, nearly 20% of Americans report difficulty swallowing oral medication over the course of a year and 3% of patients say they experience dysphagia at least once a week [Cho et al. 2015]. Dysphagia and swallowing dysfunction are also prominent in central nervous system disorders such as dementia, Parkinson’s disease, and multiple sclerosis [Deal et al. 2005; Daniels, 2006]. Both GERD and PPI use are reported to be associated with difficulty swallowing [Cho et al. 2015]. An ODT alternative to a capsule may make PPI treatment easier for these patients.
In the present study comparing the pharmacokinetic and pharmacodynamic profiles of two 30 mg ODTs with one dexlansoprazole 60 mg capsule, the systemic exposure (AUC) was roughly 25% lower in participants receiving the two ODTs than in participants receiving the capsule. Similar peak dexlansoprazole concentrations (Cmax) were observed after ODT and capsule administration. On day 5, mean pH profiles after daily doses of two 30 mg ODT or one 60 mg capsule were similar; both regimens maintained intragastric pH above 4 for 60% of the 24-hour period. The pharmacokinetic profile of 60 mg dexlansoprazole administered as two ODTs or one capsule was not affected by multiple dosing, as the systemic exposure to dexlansoprazole was equivalent on days 1 and 5 for each formulation.
The reason for reduced bioavailability is unclear, but the equivalent pH control maintained after administration of two 30 mg ODTs compared with a single 60 mg capsule suggests that adequate exposure is achieved to maximize pharmacodynamic effect. Importantly, the intragastric pH profile over the 24-hour period after dosing of 60 mg dexlansoprazole was similar irrespective of ODT or capsule administration. Higher mean pH values were observed on day 5 than on day 1 for participants receiving dexlansoprazole ODT and capsule. This shift in pH after multiple daily dosing could be due to the cumulative acid-suppressive effect of PPIs. In the acidic environment of the gastric parietal cell, PPIs convert to active sulfenamides; the binding of sulfenamide to the proton pump results in acid secretion inhibition, and its prolonged binding after multiple dosing results in an accumulative inhibitory effect [Vakily et al. 2009].
With regard to dexlansoprazole’s safety profile, there were no differences in adverse events reported when either two 30 mg ODTs or one 60 mg capsule was administered, and there were no serious adverse events reported for either treatment regimen.
The findings from this study indicate that although the criterion for pharmacokinetic bioequivalence was not met due to the difference in AUC, the acid-suppressing activity of the dexlansoprazole 60 mg capsule can be achieved with administration of two dexlansoprazole 30 mg ODTs. The prescribing information for dexlansoprazole states that two 30 mg dexlansoprazole ODTs are not interchangeable with one 60 mg dexlansoprazole capsule, based on the reduced bioavailability observed with the administration of two 30 mg ODTs in this study [Takeda Pharmaceuticals America, Inc., 2016]. The safety profiles for both formulations were similar and equally well tolerated.
Footnotes
Acknowledgements
The authors thank Dr Michael Cwik, an employee of Takeda, and the staff of PPD, Middleton, WI, for conducting the bioanalytical portion of the study. The authors also thank Ms Mary Ellen Palubicki for managing the conduct of the study. All authors had access to the data and vouch for the veracity, completeness, and analysis of the data.
Funding
The clinical studies were funded by Takeda Development Center Americas, Inc. Medical writing assistance was provided by inVentiv Medical Communications and was funded by Takeda Pharmaceuticals USA, Inc.
Conflict of interest statement
All authors are employees of Takeda Development Center Americas, Inc., a wholly owned subsidiary of Takeda Pharmaceuticals America, Inc. Funding for this study was provided by Takeda Development Center Americas, Inc.