
Abstract
The incidence of oesophageal adenocarcinoma has increased dramatically in the developed world in the last half century. Over approximately the same period there has been an increase in the prevalence of obesity. Multiple epidemiological studies and meta-analyses have confirmed that obesity, especially abdominal, visceral obesity, is a risk factor for gastro-oesophageal reflux, Barrett’s oesophagus and oesophageal adenocarcinoma. Although visceral obesity enhances gastro-oesophageal reflux, the available data also show that visceral obesity increases the risk of Barrett’s oesophagus and adenocarcinoma via reflux-independent mechanisms. Several possible mechanisms could link obesity with the risk of oesophageal adenocarcinoma in addition to mechanical effects increasing reflux. These include reduced gastric Helicobacter pylori infection, altered intestinal microbiome, factors related to lifestyle, the metabolic syndrome and associated low-grade inflammation induced by obesity and the secretion of mediators by adipocytes which may directly influence the oesophageal epithelium. Of these adipocyte-derived mediators, increased leptin levels have been independently associated with progression to oesophageal adenocarcinoma and in laboratory studies leptin enhances malignant behaviours in cell lines. Adiponectin is also secreted by adipocytes and levels decline with obesity: decreased serum adiponectin levels are associated with malignant progression in Barrett’s oesophagus and experimentally adiponectin exerts anticancer effects in Barrett’s cell lines and inhibits growth factor signalling. At present there are no proven chemopreventative interventions that may reduce the incidence of obesity-associated oesophageal cancer: observational studies suggest that the combined use of a statin and aspirin or another cyclo-oxygenase inhibitor is associated with a significantly reduced cancer incidence in patients with Barrett’s oesophagus.
Introduction
Obesity has been shown to increase the risk of many health complications, including diabetes mellitus, nonalcoholic fatty liver disease, coronary heart disease, certain forms of cancer and sleep-breathing disorders [Ogden et al. 2007] (see Table 1). The prevalence of obesity has increased substantially in the developed world in recent decades. Over approximately the same period there has been a dramatic increase in the incidence of oesophageal adenocarcinoma (OAC), also predominantly in the western, developed world. The pathogenesis of OAC is not fully defined, although increasingly the molecular changes are being understood [Gibson et al. 2013]. It seems clear that reflux of gastroduodenal contents is involved in the initiation, perpetuation and progression of the oesophageal changes. However, there must be other factors also driving these changes. In most, if not all cases, OAC arises from metaplastic columnar epithelium [Barrett’s oesophagus (BO)]. Thus it is possible and indeed likely that exogenous and endogenous factors influence gastro-oesophageal reflux disease (GORD) and the body’s response to this, the development and maintenance of BO and the progression to cancer. There are epidemiological and biological data linking obesity with the development of OAC but the associations between obesity and the three closely linked oesophageal conditions are still not fully understood, in part because interpretation of the literature is confused by different studies using a variety of measures of obesity and a variety of control groups. In this review the associations between GORD, BO, OAC and obesity are examined and the possible mechanisms linking obesity to OAC discussed, with specific reference to the role of adipocytokines secreted by fat cells.
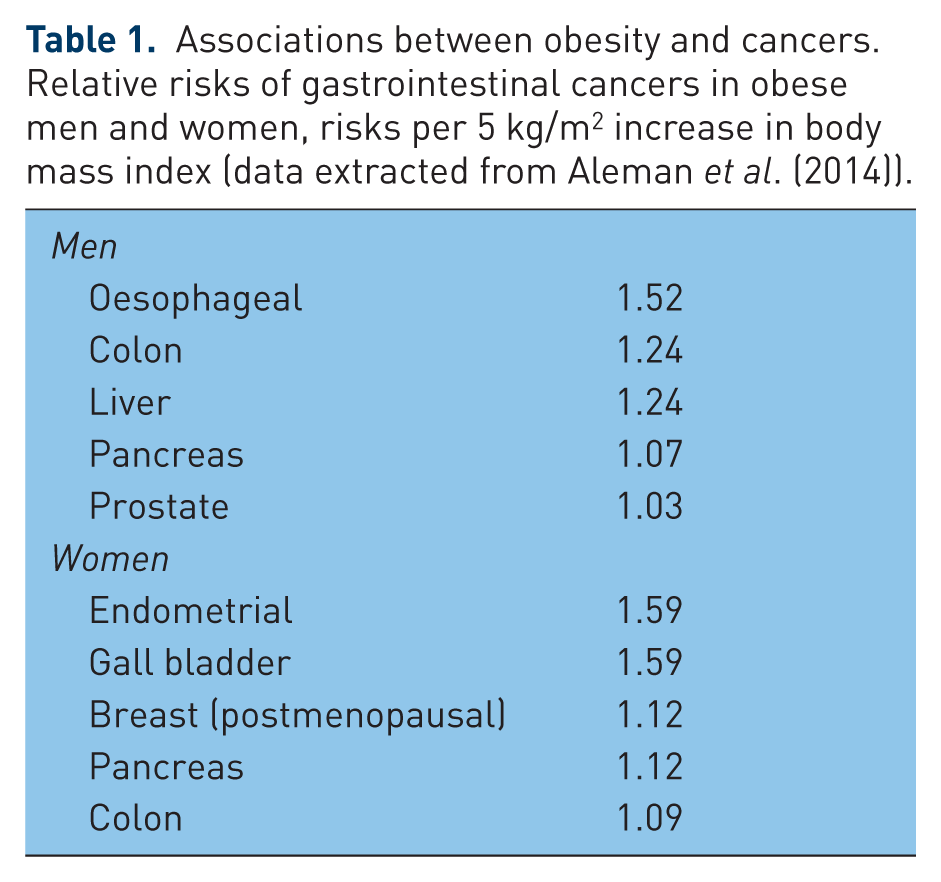
Associations between obesity and cancers. Relative risks of gastrointestinal cancers in obese men and women, risks per 5 kg/m2 increase in body mass index (data extracted from Aleman et al. (2014)).
Prevalence of obesity
Over the last half of the twentieth century obesity rates have been steadily increasing, with the highest prevalence in the USA and western Europe, although with different rates in different countries [Nguyen and El-Serag, 2010]. However, prevalence is also increasing in lower and middle income countries, making it a worldwide epidemic [Popkin et al. 2012]. The World Health Organization (WHO) estimates that 12% of the world’s population over 20 years old is now obese, which amounts to 500 million adults. This includes 10% of men and 14% of women, which has doubled from the 1980s when 5% of men and 8% of women were obese [Popkin et al. 2012]. WHO measures obesity by a body mass index (BMI) greater than or equal to 30 kg/m2. However, BMI is a crude measure of obesity as it does not take into account the composition of body mass or distribution of fat in the body. There are many other measures of obesity, such as waist circumference (WC), measuring predominantly abdominal fat, or more accurate and objective measures such as assessment of visceral fat using computed tomography (CT) or magnetic resonance imaging and these measures may be more specific to the oesophageal pathologies. However, the majority of studies have used BMI, and this does have the advantage of simplicity and ease of comparison.
Obesity and GORD
The incidence of GORD has increased worldwide; rates are highest in North America (27.8%) and lowest in East Asia (7.8%) [El-Serag et al. 2013]. Data on the association between obesity and GORD have been controversial and studies are complicated by different measures of GORD with symptoms, endoscopy and pH monitoring all being used in different studies; symptoms and even endoscopy correlates poorly with abnormal oesophageal acid exposure. For example, the sensitivity of symptoms to predict erosive oesophagitis has a sensitivity of only 30–76% and a specificity 62–96% [Katz et al. 2013]. In addition, a variety of study designs have been utilized using different comparators, such as gastroscopy controls, colonoscopy controls and population controls, and this may have contributed to some of the variability in results.
A large cross-sectional study that found no statistical association measured weight by recall of patients from 20 years beforehand, which may explain why no association was found [Lagergren et al. 2000]. However, other studies have also failed to find an association between BMI and GORD [Solhpour et al. 2008; Fujikawa et al. 2012]. Other studies have found a positive association but found it to be more robust or only positive in women [Zheng et al. 2007; Piretta et al. 2007; Breckan et al. 2009].
Many more studies have found an association between GORD and BMI [Nocon et al. 2006; Aro et al. 2005; Dore et al. 2008; Ebrahimi-Mameghani et al. 2008]. A meta-analysis including data from 18,346 patients showed a positive statistical association, even when stratified for country and level of BMI, with obesity having an odds ratio (OR) of 2.15 [Corley and Kubo, 2006] for GORD. Another meta-analysis found a consistent statistical association, with patients with a BMI greater than 30 having twice the risk of GORD compared with those with a normal BMI [Hampel et al. 2005].
A recent cross-sectional study that included 1580 participants found that compared with those with a BMI less than 25 there was a 90% increase in prevalence of GORD in those with a BMI greater than 35 [OR 1.89; 95% confidence interval (CI) 1.13–3.16]. This increase was not significantly different between sexes [Pandeya et al. 2012].
Increases in BMI, even when within normal range, are associated with worsening symptoms [Jacobson et al. 2006]. Each separate rise in BMI unit increases the risk of GORD by 4% [Kulig et al. 2004] with a dose-dependent effect [Jacobson et al. 2006]. Symptoms themselves increase in severity and frequency with increasing BMI [Nocon et al. 2007; Peura et al. 2013]. Increasing BMI is also a risk factor for developing GORD if originally symptom free at baseline [Ford et al. 2013]. In a follow-up study of patients with symptomatic GORD, increased BMI was also a major predictor of remaining symptomatic after 5 years [Azumi et al. 2008]. A 20-year longitudinal cohort follow up found that those who were obese had double the number of hospitalizations due to GORD than those in the lowest BMI quartile [Ruhl and Everhart, 1999]. This positive association has been reported globally [Moraes-Filho et al. 2005; Sakaguchi et al. 2008; Ma et al. 2009; Murao et al. 2011; Ben Chaabane et al. 2012], although the association does display some ethnic variation [Niu et al. 2012]. The association is also true in children [Pashankar et al. 2009].
As well as self-reported symptoms, increased acid reflux, as measured by oesophageal physiological monitoring, is also associated with increasing BMI [Ayazi et al. 2009]. A cross-sectional study of 223 patients found a significant nonlinear relationship between oesophageal acid exposure and increasing BMI with acid exposure, increasing most rapidly in the overweight and plateauing in the obese [Aslam et al. 2012]. Increasing episodes of reflux were also more frequent in patients with obesity when they were recumbent, suggesting obesity causes dysfunction of the oesophagogastric junction [Hajar et al. 2012].
Studies looking at the impact of weight loss on the prevalence of GORD support an important association, as rates of GORD significantly decreased from 37% to 15% with an average weight loss of 13 ± 7.7 kg [Singh et al. 2013a]. Results from the HUNT study also found a reduction in weight improved symptoms in a dose-dependent manner [Ness-Jensen et al. 2013]. A systematic review found that improvement of symptoms was most noted when treated with a gastric bypass [De Groot et al. 2009].
Location of the fat, such as abdominal obesity, rather than excess weight has been suggested as the true association of the increase in GORD. The association between BMI and GORD was attenuated when adjusted for WC, suggesting BMI has its affect by increasing abdominal obesity [El-Serag et al. 2007].
Although a large cross-sectional study in China found no association between GORD and WC or waist to hip ratio (WHR) [Chen et al. 2012], the majority of research supports the association of an enlarged WC increasing the risk of erosive oesophagitis [Kang et al. 2007; Ha et al. 2010; Tai et al. 2010]. Analysis of a cohort of 5329 Korean individuals showed an association between abdominal visceral adipose tissue (VAT) volume, but not BMI or WC, and erosive esophagitis [Nam et al. 2010]. CT scans showing high levels of VAT found this was significantly associated with duration of GORD symptoms [El-Serag et al. 2014]. The association is most obvious in the white population, which could help explain the high levels in the developed world. It is not associated with black or Asian ethnicities [Corley et al. 2007b]. In a recent meta-analysis including 19 studies, patients with central obesity had a higher risk of erosive oesophagitis [adjusted OR (aOR) 1.87; 95% CI 1.51–2.31] compared with those with normal body habitus [Singh et al. 2013b].
A cross-sectional study of 8936 participants supports the hypothesis that obesity increases the amount of acid reflux, as those with chronic atrophic gastritis are significantly less affected [Gao et al. 2010].
Obesity and BO
BO is an acquired metaplastic change from squamous epithelium to stratified columnar epithelium with goblet cells. It is a precursor lesion with potential for neoplastic change. The prevalence of BO has been increasing continuously in the Netherlands [van Soest et al. 2005], with a currently predicted prevalence of 1.6% in the general population of northern Sweden [Ronkainen et al. 2005]. The development of BO has been linked to longstanding GORD, male sex, increasing age and history of smoking [Edelstein et al. 2009]. There may be a hereditary element but it has been estimated that this only accounts for 7.3% of cases [Chak et al. 2006].
Inconsistent findings have been found regarding association of BMI with BO, some studies finding no association [Gerson et al. 2007; Corley et al. 2007a; Masci et al. 2011], some finding an association only in women [Jacobson et al. 2009; Steevens et al. 2001], and some finding a positive association [Stein et al. 2005; Smith et al. 2005; Bu et al. 2006; Anderson et al. 2007]. A meta-analysis found that BMI is not associated with BO [Cook et al. 2008] and instead it is WC that confers a twofold risk for BO [Kramer et al. 2013]. A large case–control study of 2502 participants (1102 cases and 1400 controls) found that when adjusted for WC, BMI lost its inconsistent positive association. When WC was adjusted for BMI it was still significantly associated with BO in men (OR 2.24, 95% CI 1.08–4.65) and women (OR 3.75, 95% CI 1.47–9.56). When analysed continuously there was a significant positive trend in men and women. WHR in the lowest quartile had half the risk of BO than the highest quartile in men (OR 0.44, 95% CI 0.29–0.67), although this relationship was not seen in women [Kubo et al. 2013]. This suggests that WC but not BMI is an independent risk factor for BO. Other studies supported this finding and also found an inverse relationship between BO and gluteofemoral obesity [Edelstein et al. 2007; Rubenstein et al. 2014b]. This could be due to the less metabolically active nature of gluteofemoral adipose tissue, which further supports the theory it is distribution of adipose tissue not just overall increase in weight or the excess fat tissue that is a risk for BO.
Although WC increased the risk of BO in both men and women, the association between women is attenuated when adjusted for GORD symptoms, but not in men [Kendall et al. 2013]. This suggests that non-GORD-related mechanisms contribute more to development of BO in men and these extra mechanisms could explain the higher male prevalence of BO.
However, WC is a nonspecific measurement of location of adipose tissue which can be stored subcutaneously or viscerally. SAT contributes to mechanical pressure on the stomach and oesophagus but VAT is also metabolically active. A recent case–control study of 693 patients (176 with BO, 344 colonoscopy controls and 173 endoscopy controls) used CT scans to assess VAT versus SAT. VAT was 43 cm2 larger in patients with BO than the colonoscopy control group (p = 0.006). The ratio of VAT/SAT was also significantly greater in the BO group compared with the colonoscopy control group, with the highest tertile having over a twofold increased risk of BO. The association was particularly strong in white men and those with a segment of BO greater than 3 cm in length. However, the association was not as strong when compared with the endoscopy control group, probably due to the high levels of GORD in this population [El-Serag et al. 2014]. Smaller studies support these findings that it is most specifically visceral rather than total abdominal obesity that confers the greatest risk of BO [Akiyama et al. 2009]. A recent meta-analysis [Singh et al. 2013b] included 40 studies and reported that central obesity was associated with a higher risk of BO (aOR 1.98; 95% CI 1.52–2.57). This association persisted after adjusting for BMI (aOR 1.88; 95% CI 1.20–2.95). In studies that used a control group of GORD subjects or adjusted for GORD symptoms, the association was maintained (aOR 2.04; 95% CI 1.44–2.90).
Obesity and OAC
Oesophageal cancer is the eighth most common cancer worldwide. The incidence of OAC has increased rapidly from 6.2 per 100,000 men in 1995–97 to 9.4 per 100,000 in 2008–10 [Schlansky et al. 2006; Edgren et al. 2013]. The precursor lesion BO and risk factor GORD have also risen in prevalence, making it the most dominant histological subtype of oesophageal cancers in the western developed world [Schlansky et al. 2006]. The association with obesity could offer a potential modifiable lifestyle risk factor to help reduce this incidence.
Studies in the mid 1990s first suggested the associations between obesity and OAC [D’Avanzo et al. 1996; Vaughan et al. 1995; Brown et al. 1995]. A study of 2 million Norwegians gave a relative risk (RR) = 1.80 in men who were overweight (BMI 25–29 kg/m2) and RR = 2.58 in men who were obese (BMI ≥ 30 kg/m2) [Engeland et al. 2004]. Another large cohort study of 120,852 participants found that obesity gave a RR = 4.0 (95% CI 2.3–6.9) compared with normal weight [Merry et al. 2007]. A study of 480,475 participants found a hazard ratio (HR) of 2.27 (1.44–3.59) for a BMI greater than 35 kg/m2 [Abnet et al. 2008]; other studies have reinforced this positive association between BMI and OAC [Lindblad et al. 2005; Whiteman et al. 2008].
A meta-analysis of 22 studies showed a nearly threefold increase in risk with a BMI greater than 30 kg/m2 (RR = 2.73) and that for every 5 kg/m2 increase in BMI, there was a RR of OAC = 1.11. This association was the same for gender and geographical location [Turati et al. 2013]. This linear increase in risk with BMI was not affected when stratified for GORD symptoms and when analyzed together the increased risk was more than additive [Hoyo et al. 2012]. It has been estimated that high BMI accounts for 23% of OAC, with GORD accounting for a further 36% [Olsen et al. 2011]. The high prevalence of both obesity and GORD in combination could contribute increasing incidence of OAC in many areas of the developed world. Sweden, where there are lower rates of obesity and GORD, has a lower incidence of OAC [Lofdahl et al. 2011]. The association between obesity and OAC is stronger in younger participants [Chow et al. 1998]. Obesity significantly lowers the mean age of diagnosis of OAC, from 63.6 in those without obesity to 58.9 years in those with obesity [Chak et al. 2009]. In contrast, Kong and colleagues, using data from the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) database, developed a disease simulation model and estimated that only 6–8% of the increase in OAC cases since 1973 could be directly attributed to the rise in obesity [Kong et al. 2011].
Such a robust association has led to the investigation of the effect of different fat distributions. A large prospective study of 346,554 participants looked at a variety of anthropometric measures (BMI, WC and WHR). All were positively associated with OAC, with an increase of 1 kg/m2 BMI increasing risk by 1.08 fold (95% CI 1.02–1.14), a 5 cm higher WC increasing risk by 1.16 fold (95% CI 1.04–1.29) or a 0.1 unit higher WHR increasing risk by 1.59 fold (95% CI 1.12–2.26) [Steffen et al. 2009]. The WC association with OAC was not attenuated when adjusted for BMI, adding to the evidence that abdominal fat is more important than just excess weight [O’Doherty et al. 2012; MacInnis et al. 2006; Corley et al. 2007a]. Patients with OAC were also found to have significantly more VAT on CT compared with controls [Beddy et al. 2010]. In a meta-analysis including data from six studies, when compared with normal body habitus, central adiposity was associated with higher risk of OAC (aOR 2.51; 95% CI 1.54–4.06) [Singh et al. 2013b].
Studies looking at the impact of obesity on survival have found differing results. One study of 796 patients found that obesity in never smokers shortened disease-specific survival twofold [Yoon et al. 2011]. Other studies have found no significant difference in survival in patients with obesity [Madani et al. 2010; Shridhar et al. 2012].
Mechanisms of the link between obesity of OAC
There seems little doubt that obesity, specifically visceral obesity, is a risk factor for OAC, although exactly how obesity interacts with the progression from normal oesophageal mucosa through nondysplastic Barrett’s change, dysplasia, carcinoma and the later stages of invasion and metastasis remains to be fully clarified (see Table 2). Similarly, exactly whether obesity provokes inflammatory oesophagitis and whether this directly influences neoplastic progression remains unclear. It seems very likely that multiple mechanisms are involved, possibly interacting at different stages of malignant progression. There are several, not mutually exclusive, links that could relate obesity to OAC:
They are not linked mechanistically and the association is coincidental.
A separate factor provokes both obesity and OAC.
There are confounding factors associated with obesity that promote OAC.
The mechanical effects of obesity provoke GORD and drive neoplastic progression.
Low-grade systemic inflammation associated with obesity generates a procarcinogenic environment.
The fat tissue promotes progression along the Barrett’s–dysplasia–cancer sequence by indirect, metabolic or endocrine effects.
Epidemiological links between obesity and processes involved in oesophageal adenocarcinoma development.

BMI, body mass index; CT, computed tomography; GORD, gastro-oesophageal reflux disease.
It is possible that the apparent link between obesity and OAC is nothing more than a chance association. Although overall there have generally been parallel increases in obesity and OAC in the last few decades, and epidemiologically, obesity (especially abdominal visceral obesity) is clearly a risk factor for BO and OAC [Singh et al. 2013b], there are enough striking inconsistencies to at least question a direct link. There have been dramatic rises in OAC incidence in Australia and Denmark with much more modest changes in obesity. Although the epidemic of OAC seen in the developed world seemed to start in the UK about 10 years before the USA, the UK was about 10 years behind the USA in the upsurge of obesity rates [Edgren et al. 2013] and as discussed previously at least one simulation model suggests the rise in obesity contributes only a small effect to the rise in OAC [Kong et al. 2011]. Another major area of contention is why does OAC occur with such a predilection for white people? Although obesity rates in the USA have increased in all racial groups (most rapidly in black people and Hispanics, least so in Asians) the rise in OAC has really been limited to white people [Wong et al. 2014]. This suggests that more than simply obesity is responsible but whether this reflects underlying genetic predisposition, physiological and metabolic differences or other factors requires further analysis. However, it is notable that the link between visceral obesity and the development of BO seems most pronounced in white men [El-Serag et al. 2014].
The majority of the epidemiology showing obesity as a risk factor of OAC is quite compelling; obesity is associated with the risk of many other cancers (with biological plausibility) and the relative risk of OAC with obesity is higher than other cancers, all suggesting that the association is causal even if obesity is not the sole driver of OAC [Aleman et al. 2014] (Table 1).
Association with Helicobacter pylori and gastric ghrelin
Infection with Helicobacter pylori, particularly the CagA-positive strains that provoke more intense gastric mucosal inflammation, has been shown to be inversely associated with erosive oesophagitis and BO [Rubenstein et al. 2014a; Garcia et al. 2014]. The most plausible explanation for this is that infection of the gastric body with H. pylori reduces gastric acid secretion by either reversible means due to local cytokine production [Beales and Calam, 1998, 2001] or more irreversibly due to the development of gastric atrophy [el Omar et al. 1997]. Thus active H. pylori infection would be associated with less reflux disease. Over the last century the prevalence of H. pylori infection has fallen, whilst the incidence of OAC has increased [Banatvala et al. 1993]. Weight gain is common after H. pylori eradication and hence a reduced prevalence of H. pylori could directly provoke more severe reflux disease and an overall increase in body mass. A direct link between these two has been postulated via the role of gastric ghrelin. Ghrelin is a peptide hormone produced in, and secreted from, the gastric body that stimulates appetite. H. pylori infection is associated with lower levels of gastric mucosa ghrelin and mucosal levels increase with eradication. Hence it has been postulated that lower levels of H. pylori infection are directly linked to obesity by increasing appetite. Whilst this is an attractive hypothesis, serum ghrelin levels have not been shown to reliably increase after H. pylori eradication and higher plasma ghrelin levels seem to associated with a lower incidence of erosive oesophagitis (possibly by enhancing gastric emptying), suggesting that increased levels of ghrelin are more likely to have inhibitory effects on OAC development [Boltin and Niv, 2012]. To confuse this issue further, increased ghrelin levels have also been positively associated with the development of BO [Rubenstein et al. 2013].
Birth cohort studies have shown that the prevalence of H. pylori had been falling steadily throughout the twentieth century and not obviously related to the increase in OAC [Banatvala et al. 1993] and whilst the upsurge in OAC incidence in Sweden seemed to begin in the early 1990s, after the discovery appreciation of the pathogenesis of H. pylori, the upsurge in OAC in the UK began in the 1960s, well before the discovery of H. pylori and any effects of widespread active eradication of H. pylori. Hypothesizing that decreased gastric H. pylori infection as a direct cause of both obesity and OAC is also unable to explain the clear gender and racial differences in OAC [Edgren et al. 2013].
The role of the intestinal microbiome
Our increasing understanding of the complex interrelationships between humans and their microbiome are beginning to suggest other ways in which factors may influence both obesity and OAC either separately or via common and linking pathways. The intestinal microbiome is significantly different in patients with obesity compared with those of normal weight [de Vos and de Vos, 2012] and the microbiome changes with weight loss [Ley et al. 2006]. The intestinal microbiome appears to actually influence systemic metabolism, weight gain, systemic inflammation and the complications of obesity [Niewdorp et al. 2014]. Weight gain has been transmitted by faeces in experimental mice [Ridaura et al. 2013].
In addition to the direct links involving changes in acid secretion already mentioned, it is possible that clearance of gastric H. pylori alters the gastric microbiome in other as yet uncharacterized ways that could promote appetite and obesity, increase acid reflux, impair gastric emptying or alter mucosal immune responses in ways that enhance distal oesophageal carcinogenesis [Abreu and Peek, 2014; Calatayud et al. 2002].
Intriguing data have shown that the normal, intact oesophagus has a distinctly different microbiome to the damaged oesophagus (erosive oesophagitis or BO). The normal oesophageal microbiome is predominantly gram-positive organisms and the abnormal oesophagus dominated by gram negatives [Yang et al. 2009]. It not yet known if this is cause or effect, or how this relates to obesity. It is postulated that the abnormal oesophageal microbiome may contribute to acid reflux, inflammation and neoplastic progression by the local production of mediators, such as lipopolysaccharide, that activate inflammatory responses and increase nitric oxide production, which in turn may exacerbate reflux disease and promote the development of OAC [Yang et al. 2009; Abreu and Peek, 2014; Fan et al. 2001]. Therefore it is possible, although somewhat speculative, that the intestinal microbiome changes are separately enhanced by obesity and OAC. It is clear that the microbiome has profound effects on many aspects of human physiology and further studies will likely reveal if this is a link between OAC and obesity.
Association with lifestyle factors
The aetiology of obesity is complex and multifactorial. It is possible that specific dietary or lifestyle factors associated with obesity promote OAC development. For example, regular physical exercise is associated with a lower rate of erosive oesophagitis and protects against obesity [Falk et al. 2011]. Smoking is a risk factor for both BO and progression to OAC [Pohl et al. 2013; Cook et al. 2012] and has a complex relationship with body weight. The lifestyle associated with significant obesity may be associated with greater proclivity to smoking, but smoking is also associated with lower body weight. The overall prevalence of smoking has declined over the same period that OAC has increased, suggesting that this is not the predominant mechanism of carcinogenesis.
Several dietary substances promote obesity and relaxation of the lower oesophageal sphincter (LOS), possibly promoting reflux disease and OAC. For example, chocolate promotes LOS opening and is more commonly consumed by people with obesity [Falk et al. 2011]. There are many other putative dietary components that could directly or indirectly promote OAC development in people with obesity, for example OAC is also associated with increased meat intake and reduced fruit and vegetable intake [Navarro Silvera et al. 2011]. At present there are no robust data implicating any particular agent. Although use of vasodilator agents that may also relax the LOS to treat hypertension, angina and heart failure has also increased over the last few decades, and these drugs are likely to be disproportionately prescribed to people who are overweight, a meta-analysis has shown no association between these drugs and OAC [Alexandre et al. 2012].
Mechanical effects of obesity
As discussed previously, it is increasingly recognised that the link between OAC and fat tissue is much stronger for visceral obesity than overall obesity [Duggan et al. 2013]. Perhaps the most obvious pathogenic link is that the visceral fat tissue exerts mechanical effects on the upper gastrointestinal (GI) tract to promote GORD directly and the Barrett’s–cancer sequence indirectly. There are some data to support this: obesity is associated with greater oesophageal acid exposure [El-Serag et al. 2007], increased size of hiatus hernia [Pandolfino et al. 2006] and increased transient lower oesophageal relaxations [Wu et al. 2007].
Several recent sophisticated studies have examined the mechanical effects of visceral obesity on the physiology of the gastro-oesophageal junction. Even in asymptomatic healthy volunteers, an increased WC was associated with an increased length of inflammation in the mucosal segment between squamous oesophageal mucosa and gastric oxyntic mucosa [Robertson et al. 2013]. The increased WC was also associated with increased depth of intrasphincteric acid reflux (reflux within the LOS without fully traversing it) and shorter functional LOS [Robertson et al. 2013]. In addition, increasing WC was associated with lower postprandial peak LOS pressure, displacement of the squamocolumnar junction proximally, and blunting of the physiological proximal movement of the squamocolumnar junction during transient LOS relaxations (TLOSRs). These findings have been interpreted as indicating the development of a partial hiatus hernia. These changes were not associated with increased acid reflux measured by conventional means (at 5 cm above the LOS) but were associated with increased acid reflux just proximal to the gastro-oesophageal junction (0.5 cm). Although TLOSRs were not increased in the obese group, the proportion of TLOSRs carrying acid was significantly increased (32.8% compared with 4.2%). Similar changes can be reproduced by wearing a waist belt, suggesting these are manifestations of a mechanical effect of visceral fat [Lee at al. 2013]. The exact means of the mechanical effects are unclear but could involve abdominal pressure pulling open the distal LOS, mechanical deformity of the area preventing locally secreted gastric acid from freely flowing away from the area of reflux or proximal displacement of the gastro-oesophageal junction moving the acid-secreting gastric mucosa proximally into a zone of higher pressure, and so promoting reflux [Robertson et al. 2013].
These mechanical effects of abdominal fat may provoke both typical gastro-oesophageal acid reflux and more localized distal or even intrasphincteric reflux, and might contribute to the development of adenocarcinoma. However, there are considerable data showing that obesity is associated with BO or OAC independently of measures of reflux, so it seems likely that visceral fat tissue exerts both direct and indirect effects on the promotion of oesophageal carcinogenesis [El-Serag et al. 2014; Garcia et al. 2014; Lagergren et al. 1999].
Metabolic actions of adipose tissue
Once regarded as a rather inert, storage organ, it is now recognized that adipose tissue is a complex metabolically active tissue, which secretes a variety of mediators that can have effects throughout the body. It is perhaps easiest to consider these secretions in two groups: those specific (at least relatively so) for adipose tissue, generally called adipokines or adipocytokines, which include several important mediators including leptin, adiponectin, resistin and omentin; and a second group of systemic cytokines that can be produced by a variety of tissues, including fat cells [Yeung et al. 2013; Donohoe et al. 2014; Aleman et al. 2014]. It is now considered that obesity is a state of chronic low-grade, sterile, systemic inflammation, due to increased numbers of activated immune cells infiltrating the fat tissues. These immune cells are responsible for a complex array of pro- and anti-inflammatory interactions, but obesity is associated with increased circulating levels of a variety of proinflammatory mediators secreted by these adipose tissue associated immune cells. These include tumour necrosis factor α (TNFα), interleukin (IL)-1β, IL-6, IL-8, interferon γ, monocyte chemoattractant protein 1 and vascular endothelial growth factor. It is believed these changes contribute not only to the development of the metabolic syndrome, insulin resistance and the related complications but also to the increased risk of many cancers associated with obesity [Yeung et al. 2013]. The complex pathogenesis of the inflammation associated with obesity is outside the scope of this review but this has recently been subjected to an excellent overview [Lee and Lee, 2014].
Although inflammation is recognized as a classical precursor to cancer, it is not completely understood how this systematic inflammatory state promotes cancer, although clearly many of these mediators promote cell proliferation, inhibit apoptosis and stimulate angiogenesis, all of which would be expected to promote cancer. An increased incidence of most GI cancers are associated with this obesity-induced inflammatory state [Aleman et al. 2014] and it has been shown that faecal calprotectin (as a marker of luminal inflammation) is increased in obesity [Kant et al. 2013]. However, it is more difficult to relate these changes specifically to OAC, which does seem to have the highest relative risk of GI cancers associated with obesity: it might be expected that the obvious and prolonged ongoing inflammation or bile acid induced proliferation induced by reflux in Barrett’s epithelium might override these low-grade inflammatory drivers and there is no clear parallel in the oesophagus to the fat-induced inflammatory changes that can precede liver and pancreatic cancers.
The metabolic syndrome and insulin resistance
Another feature of the systemic inflammation associated with obesity is insulin resistance and increased circulating concentrations of both insulin and insulin growth factor 1 (IGF-1). This seems at least partly driven by systemic and paracrine secretions from metabolically active VAT: in visceral obesity there are increased levels of inflammatory cytokines and mediators, including free fatty acids, TNFα, leptin and resistin [Ryan et al. 2008; Eksteen et al. 2001] and reduced secretion of adiponectin [Mokrowiecka et al. 2012]. A complex array of adipose-tissue infiltrating immune cells are responsible for the majority of the cytokines produced, although it is not completely understood how this inflammation leads to insulin resistance (for a full review of this complex area, see Lee and Lee [2014]). Increased circulating insulin promotes carcinogenesis partly by stimulating the production of IGF-1 and it also downregulates production of IGF binding proteins 1 (IGFBP-1) and 3 (IGFBP-3) [Greer et al. 2012]. This leads to an increase in bioavailable IGF-1, which can bind to the IGF receptor complex, activating pathways that promote tissue proliferation. Insulin resistance has been associated with progression to adenocarcinoma in a Barrett’s cohort [Duggan et al. 2013] but overall data are conflicting on involvement of the complex insulin/IGF-1 signalling in obesity-associated OAC. Insulin and IGF-1 are mitogenic for many tissues, including Barrett’s OAC cell lines, which express IGF-1 receptors [Doyle et al. 2012] and IGF-1 receptor expression has been reported to be increased in OAC resected from patients who are viscerally obese [Donohoe et al. 2012]. The IGF-1 receptor has been reported to be expressed in 67% of OAC cases [Nagaraja and Eslisk, 2014] but using quantitative polymerase chain reaction IGF-1 receptor expression was not increased in OAC compared with normal oesophageal mucosa and there was no association between IGF-1R and BMI, tumour differentiation and stage or survival [Zhao et al. 2009].
Serum levels of IGF-1 have been reported to be higher in patients with OAC than in those with nondysplastic Barrett’s or healthy controls [Doyle et al. 2012] and, as assessed by immunohistochemistry, activation of intracellular signalling pathways downstream of insulin–IGF-1 is common in patients (> ~40%) at all stages along the Barrett’s–OAC sequence [Greer et al. 2013]. Activation was more pronounced in high-grade dysplasia and cancer than nondysplastic mucosa. However, there was no association between intracellular signalling activation and circulating insulin, IGF-1 or IGF-2
Polymorphisms in the IGF-1 receptor, specifically carriage of an A allele in the G1013A polymorphism, seem to be associated with the risk of both BO and OAC associated with obesity: G/G homozygosity at this position was not significantly associated with either BO or OAC in this obese population [MacDonald et al. 2009], although this study did not provide any data on IGF-1 receptor expression or serum insulin or IGF-1, and the confidence intervals for all groups were wide. A separate study showed that carriage of an A allele at IGF-1R G10103A was associated with increased IGF-1R only in OAC tissues [Zhao et al. 2009]. Thus there are some data to implicate genetic variability in modifying the response to and OAC cancer risk of obesity, but further studies are required to establish why this polymorphism only influences receptor expression on a background of obesity.
Data are conflicting on the role of IGF-1 and insulin as risk factors for malignant progression in BO. Some studies have shown an increase in the risk of cancer or BO [Donohoe et al. 2012; Greer et al. 2012], but other studies have failed to show any association between serum IGF-1 or IGFBP3 (the predominant serum binding protein levels) and progression of Barrett’s [Siahpush et al. 2007].
The role of the insulin–IGF-1 system requires further exploration as a link between obesity and OAC but several issues should be considered when reviewing the data. Although IGF-1 receptors are apparently expressed on OAC cells, receptor expression does not always correlate with function. Several oesophageal squamous cell lines strongly express IGF-1 receptors but these are incapable of functional signalling [Doyle et al. 2012]. Other stimulants such as leptin and acid can also activate the downstream intracellular pathways that are typically located distal to the insulin–IGF-1 receptor complex and thus detection of activation of these pathways in vivo cannot be taken as proof of activation of the IGF-1 system [Ogunwobi et al. 2006; Beales et al. 2007].
The role of adipokines
Whilst these general inflammatory changes may be important in the development of OAC, the specific role of adipokines is attracting considerable scrutiny. Of those examined in detail, leptin and adiponectin seem to provide a direct mechanistic link between obesity and progression to OAC.
Leptin is a 16 KDa protein secreted by fat cells: serum levels are proportionate to body fat mass, in keeping with leptin being an essential regulator of appetite, energy metabolism and body weight. In the vast majority of people with obesity, serum levels are significantly elevated, leptin deficiency is a very rare cause of obesity, and a degree of hypothalamic hyposensitivity to leptin seems to contribute to obesity [Vansaun, 2013]. Increased leptin levels have been shown to be an independent risk factor for many cancers, including breast, colorectal, prostate, ovarian, lung and endometrial [Garofalo and Surmacz, 2006]. Leptin levels have been shown to be an independent risk factor for the development of BO, although another study showed this effect was confined to women [Kendall et al. 2008; Garcia et al. 2014] and increased leptin levels have been shown to be an independent risk factor for progression to cancer in a cohort of patients with BO [Duggan et al. 2013]. Expression of leptin receptors has been reported in nondysplastic Barrett’s cell lines, OAC cell lines, BO and OAC [Mokrowiecka et al. 2013; Howard et al. 2010; Ogunwobi and Beales, 2008a; Ogunwobi et al. 2006]. Increased leptin receptor expression has been reported to be associated with advanced stage in OAC [Howard et al. 2010] (Table 3).
Summary of pathogenic links between adipokines and oesophageal adenocarcinoma.

Cellular effects of leptin
Leptin promotes malignant behaviour in oesophageal cell-line models. Leptin stimulates proliferation, inhibits apoptosis, and enhances migration and invasion, as well as stimulating the production of the matrix metalloproteinases (MMPs) MMP-2 and MMP-9, which are involved in invasion [Beales et al. 2013a; Ogunwobi et al. 2006]. Incubation of OAC cell lines in conditioned media from visceral adipocytes enhances MMP-9 production and in vivo MMP-9 production is associated with visceral obesity [Allott et al. 2013]. The cell signalling pathways involved in these effects have been well described [Beales et al. 2013a; Ogunwobi et al. 2006a]: binding of leptin to the full-length receptor stimulates phosphorylation of the JAK2 tyrosine kinase, which subsequently leads to activation of the extracellular signal-related kinase (ERK) and protein kinase B/Akt cascades. The NFκB pathway is also activated, predominantly via Akt activation. The p38 mitogen-activated protein (MAP) kinase pathway is also activated downstream of the leptin receptor, although this is a JAK2-independent pathway. The ERK, Akt, NFκB and p38 pathways are all essential to the proliferative and antiapoptotic effects of leptin and these cooperate in inducing cyclo-oxygenase 2 (COX-2) gene expression. The resultant prostaglandin E2 produced then appears to continue to enhance proliferation and apoptosis by transactivating the epidermal growth factor receptor (EGFR) and further activating the MAP kinase cascades with late activation of c-Jun N-terminal kinase (JNK). Leptin increases mRNA expression of the EGFR ligands transforming growth factor α and heparin binding EGF in OAC cells and immunoneutralization of these growth factors ameliorates the proliferative effects of leptin [Ogunwobi and Beales, 2008a].
STAT3 is also activated downstream of the leptin receptor and JAK2 activation, and once activated this transcription factor is also essential to the proliferation, antiapoptotic and proinvasive effects of leptin [Beales et al. 2013a].
Thus leptin seems to be able to stimulate malignant behaviour in Barrett’s cells bearing the leptin receptor and may be a direct link between obesity and progression to OAC. As discussed previously, obesity seems to promote the development of BO and OAC through reflux-dependent and -independent means, and epidemiologically the combination of obesity and reflux is a significantly greater risk than either alone or the additive effect [Duggan et al. 2013; El-Serag et al. 2014; Garcia et al. 2014; Lagergren et al. 1999]. This is reflected in further experimental data. The combination of leptin (as a model for obesity) and transient acid exposure (as a model of transient acid reflux) produced significantly increased (and synergistic) levels of cell proliferation and inhibition of apoptosis in OAC cell lines [Beales and Ogunwobi, 2007]. This combination resulted in synergistic activation of the Akt and ERK signalling pathways, although expression of leptin receptor, COX-2 expression and p38 MAP kinase and EGFR phosphorylation were not greater in the combination acid–leptin exposure group compared with the leptin-alone treated group. It has previously been shown that oesophageal acid exposure enhances MAP kinase activation and proliferation in vivo [Souza et al. 2002], thus it is quite possible that continued exposure to the high levels of leptin seen in people with obesity enhances the response of Barrett’s mucosa to even physiological acid reflux and promotes malignant change.
Leptin is also synthesized and secreted by chief cells in the gastric body and can be detected in gastric juice. The function of this luminal leptin is unclear but it may be a physiological regulator of mucosal integrity or nutrient absorption. Thus the oesophageal mucosa is potentially exposed to both circulating leptin and that in gastric refluxate. The putative role of gastric leptin needs further exploration but it is worth noting that the presence of BO has been associated with increased levels of gastric fundic leptin [Francois et al. 2008] (see Table 3).
Effects of adiponectin
Adiponectin is a 30 KDa protein that is secreted in high concentrations by adipocytes. In contrast to leptin, adiponectin secretion falls as obesity increases. The exact mechanism underling this inverse relationship between fat mass and adiponectin secretion are unclear [Li et al. 2012]. As might be expected, in general the effects of adiponectin are opposed to those of leptin and relative adiponectin deficiency has also been implicated in the pathogenesis of the metabolic syndrome and its complications, including systemic inflammation. Adiponectin is found in various forms, which have slightly different biological actions and are detected in different assays [Dalamaga et al. 2012]. This has made comparison between studies difficult. In general, low systemic adiponectin levels have been associated with an increased risk of many cancers (including breast, colorectal, prostate, endometrial, gastric) [Kelesidis et al. 2006]. Adiponectin is secreted as a full-length monomer that then forms both low-molecular and high-molecular weight oligomers. A truncated form (globular adiponectin) is also found; this is at least partly formed by breakdown of full-length adiponectin by enzymes released in inflammation [Waki et al. 2006]. There are two specific adiponectin receptors (AdipoR1 which seems to be globular adiponectin specific) and AdipoR2 which has equal affinity for globular and full-length adiponectin [Yamauchi et al. 2014].
In experimental studies adiponectin exerts antimalignant effects in Barrett’s cancer cell lines. Adiponectin inhibits leptin-induced proliferation, invasion and migration, and ameliorates the antiapoptotic effect. Using RNA interference, it was shown that these effects are mediated via the AdipoR1 and require the activation of 5’-adenosine monophosphate activated kinase (AMPK), which ultimately leads to blunting of leptin signalling via the Akt pathway [Ogunwobi and Beales, 2008d]. More recently these inhibitory effects have been shown to be mediated by the activation of the protein tyrosine phosphatase 1B (PTP1B) [Beales et al. 2013a]: adiponectin leads to both induction of PTP1B mRNA and increased protein expression and also separate stimulation of PTP1B enzyme activity. Activation of this tyrosine phosphatase inhibits signalling via the leptin receptor. Thus, experimental models provide a basis to explain how leptin, adiponectin and acid interact at the cellular level to promote either the promotion or persistence of Barrett’s epithelium or malignant behaviour in cancer cells. This potential mechanism of adiponectin is important because PTP1B, as relatively nonspecific phosphatase, would also be expected to inhibit signalling via other pathways that are believed to be important in driving malignant behaviour in Barrett’s epithelium, such as EGFR ligands, IL-6, IGF-1, insulin and bile acids [Beales and Ogunwobi, 2009; Zhang et al. 2011].
Association between adiponectin and OAC
There are data to support relative adiponectin deficiency in the promotion of OAC. AdipoR1 and AdipoR2 are expressed on both nondysplastic and neoplastic Barrett’s epithelium [Mokrowiecka et al. 2013; Ogunwobi and Beales, 2008d; Konturek et al. 2008]. Circulating adiponectin levels have been shown to be inversely associated with the risk of BO [Rubenstein et al. 2008] and erosive oesophagitis [Kato et al. 2011]. Amongst patients with GORD increased levels of low-molecular weight adiponectin and a high low-molecular weight/total adiponectin ratio have been shown to be independently associated with a reduced risk of BO [Rubenstein et al. 2009], although other studies have failed to show this inverse relationship [Kendall et al. 2008]. Low serum levels of adiponectin have been reported to be an independent risk factor for neoplastic progression in a cohort of patients with BO [Duggan et al. 2013] (see Table 3). Both leptin and adiponectin have potentially important immunostimulatory effects and indeed both inhibitory and stimulatory effects of adiponectin have been reported, which may be related to different effects of the adiponectin forms [Ogunwobi and Beales, 2006]. However, any independent effects of these adipokines on inflammatory oesophagitis have not been adequately explored as yet.
The different types of adipose tissue have different metabolic and endocrine effects. Although OAC and Barrett’s are most clearly associated with abdominal rather than general obesity [El-Serag et al. 2014], even within abdominal obesity there are different contributions from visceral and subcutaneous fat tissues: excess visceral fat being specifically associated with BO. Gluteofemoral fat (‘hips’) appears to not be a specific risk factor of BO and may even be protective [Rubenstein et al. 2014b]. It is thought that fat in this distribution is even less metabolically active and has less effect on progression of BO. In light of this, it is important to recognize that that visceral, rather than subcutaneous, fat is usually the predominant source of circulating adiponectin [Motoshima et al. 2002; Lysaght et al. 2011; Snijder et al. 2004] and reduction in adiponectin secretion from visceral fat probably specifically contributes to the Barrett’s–carcinoma sequence.
The mechanisms linking obesity and OAC are complex and likely multifactorial, and are likely to differ depending on the phenotypic stage of the oesophageal mucosa (along the normal–oesophagitis–Barrett’s–dysplasia–cancer spectrum). The experimental and epidemiological data support a role of the adipokines leptin and adiponectin in the progression to OAC but further mechanistic and clinical studies are still required. Whilst mechanical effects due to visceral adiposity seem to enhance reflux, there do seem to be reflux-independent effects promoting both the development of BO and neoplastic progression, and biological plausibility and clinical data implicating direct roles for mediators directly secreted by adipocytes.
Implications for therapy
The fact that obesity is a risk factor for both BO and OAC is established. This is already being translated into the clinical arena: for example, the British Society of Gastroenterology guidelines now suggest that screening and case finding for BO be considered in those with chronic reflux symptoms and at least three other risk factors (age > 50, male sex, white race, obesity), this has the advantage of detecting premalignant cases of BO that may be amenable to surveillance and endoscopic therapies if required [Fitzgerald et al. 2014]. A broader question is which therapies may be applicable on a population or individual scale to reduce the incidence of OAC when associated with obesity. Obviously weight loss with dietary and behavioural modifications remains the first-line approach. Proton pump inhibitors (PPIs) are widely used for symptomatic treatment at all stages of GORD: it remains unclear whether acid suppression by these means has a genuine chemopreventative action [Fitzgerald et al. 2014] and this may not be clarified until the large UK AspECT trial, that includes arms with standard and very high-dose esomeprazole, has reported [Das et al. 2009]. The recent British Society of Gastroenterology guidelines advocate acid suppressive therapy for symptom control in patients with BO, but do not support acid suppression either via drugs or antireflux surgery purely to prevent neoplastic progression [Fitzgerald et al. 2014]. It remains controversial whether the response to PPIs is affected by body fat. Obesity (BMI > 30 kg/m2) has been shown to be significantly associated (OR 3.82; 95% CI 1.45–10.09) with failure of PPI therapy to control reflux symptoms [Dickman et al. 2011]. However, Peura and colleagues found that BMI did not negatively impact either symptom control or healing of erosive oesophagitis [Peura et al. 2013]. At present it seems appropriate to advocate PPI therapy in sufficient doses to control reflux symptoms in all patients.
For those with reflux symptoms and significant obesity, a Roux-en-Y gastric bypass appears to be the preferable procedure, if bariatric surgery is being considered, producing beneficial metabolic effects, including increased serum adiponectin levels and a reduction in body mass and visceral fat, as well as reducing symptoms from GORD, although this cannot be recommended purely for cancer prevention [Umeda et al. 2013; De Groot et al. 2009].
There may yet be some developments in therapies aimed to improve the metabolic/endocrine profile of adipose tissue. CB1 antagonists, such as rimonabant, reduce visceral fat [Despres et al. 2009] and PPARγ agonists such as rosiglitazone enhance adiponectin release from visceral fat [Motoshima et al. 2002]. However, at this time the side-effect profiles, psychiatric problems with rimonabant and bladder cancer and increased cardiovascular mortality with PPARγ agonists preclude their wider use. Other agents that may usefully increase adiponectin levels (such a PPARγ agonists, inhibitors of the renin–angiotensin system, calcium channel modulators and some β-receptor antagonists and various natural substances) deserve further study, although at present data are limited and have not been applied to oesophageal disease [Lim et al. 2014]. Adiponectin analogues [Otvos et al. 2011] and leptin receptor antagonists [Rene Gonzalez et al. 2009] are in preclinical development but these appear some way off clinical use.
The potential role of metformin deserves further exploration as a chemopreventative agent. In Barrett’s cell-line studies, the inhibitory effects of adiponectin are mediated via activation of AMPK [Ogunwobi and Beales, 2008d]. Metformin is approved for the treatment of type 2 diabetes mellitus and is the agent of first choice when associated with obesity. Metformin has pleiotropic cellular actions, including direction activation of AMPK kinase and exerts potentially useful anticancer effects, which have be demonstrated in experimental models and epidemiological studies [Martin-Castillo et al. 2010; Rizos and Eliasf, 2013]. We have confirmed similar growth and invasion inhibition in Barrett’s cell lines (Garcia-Morales and Beales, unpublished observations). Although many of the epidemiological studies suggesting chemopreventative effects of metformin are confounded by the increased risk of cancer in patients with diabetes, as well as by age, disease duration and disease severity, and also the comparison with other antidiabetic agents (the incidence of cancer may be increased with sulphonylurea use) [Franciosi et al. 2013], metformin has been associated with a reduced incidence of all cancers and associated mortality [Thakkar et al. 2013; Zhang et al. 2013]. More specifically, metformin has been associated with a reduced incidence of oesophageal cancer (OR 0.90; 95% CI 0.83–0.98) [Franciosi et al. 2013] but this was not confirmed in another study from the UK General Practice Research database (OR 1.23; 95% CI 0.92–1.65) [Becker et al. 2013b], although the latter studies did not specifically examine just adenocarcinoma. It is clear that the data relating metformin to oesophageal cancer are considerably more limited than other common cancers. Metformin has a low incidence of side effects and could be a promising chemopreventative agent, although more studies specifically in OAC are needed.
This leaves aspirin and statins (HMG-coenzyme A reductase inhibitors) as the most appropriate agents to attempt to reduce the incidence of OAC associated with, or indeed without, obesity. A considerable amount of experimental data show that COX inhibitors, such as aspirin, reduce malignant behaviour such as proliferation and also induce apoptosis in OAC and non-neoplastic Barrett’s cell lines, and nonspecific COX and COX-2 selective inhibitors block the effects of leptin in cell-line models [Ogunwobi and Beales, 2008b; Fang et al. 2011; Beales and Ogunwobi, 2010]. Although more definitive conclusions on the preventative effects of aspirin may have to wait until the AspECT trial has reported [Das et al. 2009], in observational studies and meta-analyses aspirin use has been reported to be associated with a reduced incidence of both BO and OAC [Omer et al. 2012; Beales et al. 2013]. Similarly, statins exert potent anticancer effects in OAC cell-line models by inhibiting small signalling G-protein prenylation and limiting procarcinogenic signalling from growth factor receptors [Ogunwobi and Beales, 2008c]. In experimental models the effects of COX pathway inhibition (whether using small molecule COX inhibition, microsomal prostaglandin E synthase-1 (PGES-1) inhibition or RNA interference) and statins were additive [Fang et al. 2011; Ogunwobi and Beales, 2008c] and a similar magnitude of reduced risk with combined therapy has been reported in two separate meta-analyses of Barrett’s cohorts, in which the combination of COX inhibitor and statin was associated with a 85% reduction in OAC incidence [Singh et al. 2013c; Beales et al. 2013b]. Statins may also increase serum adiponectin levels [Lim et al. 2014]. Whilst it may be premature to advocate aspirin and statin therapy as primary preventative therapy for all, it does seem prudent to ensure, at the very least that these agents are always used if appropriate for reduction of cardiovascular risk, remembering that cardiovascular disease is the main cause of mortality in the Barrett’s population [Fang et al. 2011].
Conclusion
The prevalence of obesity has increased significantly in most areas of the developed world in recent decades. There has also been a striking increase in the incidence of OAC. Although the geographical, racial, sex and time courses associating OAC and obesity are not always congruent, there are a significant amount of data showing that obesity, especially visceral abdominal obesity, is an important risk factor for BO and adenocarcinoma. Obesity does seem to provoke GORD but the positive associations between measures of obesity and BO and OAC are maintained after correction for reflux. There are several potential reflux-independent mechanisms by which obesity could promote the development of OAC. The adipokines leptin and adiponectin are secreted by visceral fat cells and there are epidemiological data implicating both relative adiponectin deficiency and increased leptin as risk factors for progression to OAC. In laboratory models leptin enhances and adiponectin inhibits malignant behaviour in Barrett’s cell lines, suggesting these mediators may have a direct role in pathogenesis. At present, there are no specific chemopreventative strategies, but appropriate weight loss in people with obesity seems appropriate. Aspirin and statins may have chemopreventative actions and should be utilized as indicated by the cardiovascular risk profile.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.