
Abstract
Sickle cell intrahepatic cholestasis is a relatively uncommon complication of homozygous sickle cell anemia, which may lead to acute hepatic failure and death. Treatment is mainly supportive, but exchange transfusion is used as salvage therapy in life threatening situations. We describe a case of a 16-year-old female with homozygous sickle cell anemia who presented to the emergency room with fatigue, malaise, dark urine, lower back pain, scleral icterus and jaundice. She was found to have marked hyperbilirubinemia, which persisted after exchange transfusion. Because of the concomitant presence of gallstones and choledocholithiasis, the patient underwent endoscopic ultrasound and laparoscopic cholecystectomy followed by endoscopic retrograde cholangiography and sphincterotomy.
Keywords
Introduction
Sickle cell trait and homozygous disease affects 1 in 10 and 1 in 600 African-Americans, respectively [Baichi et al. 2005]. Patients with sickle cell disease (SCD) can manifest a diverse spectrum of hepatobiliary diseases, from acute viral hepatitis, cholecystitis and choledocholithiasis to acute sickle hepatic crisis (ASHC), hepatic sequestration crisis, and sickle cell intrahepatic cholestasis (SCIC) [Schubert, 1986]. Sickle cell hepatopathy comprises a diverse constellation of syndromes occurring predominantly in patients with SCD and less frequently with sickle cell trait, sickle cell hemoglobin C disease and hemoglobin S β thalassemia [Banerjee et al. 2001]. ASHC results from trapping of sickled red blood cells (RBC) in the liver sinusoids and subsequent anoxic injury. Acute end organ failure rarely occurs in ASHC. In ASHC, red cells are clustered in the liver and hepatomegaly, right upper quadrant pain, and drop in hematocrit ensue. Occasionally, hematocrit improves after resolution of the sequestration crisis and reinstatement of normal liver size, indicating that not all RBC are destroyed in the hepatic sinusoids [Hatton et al. 1985]. SCIC is a rare complication that manifests with sudden onset of right upper quadrant (RUQ) abdominal pain, significantly elevated liver enzymes and hyperbilirubinemia, coagulopathy, progressive hepatomegaly, renal insufficiency, and acute liver failure [Irizarry et al. 2006; Bandyopadhyay et al. 2008]. SCIC portends a relatively high mortality risk (up to 40%) which has decreased with the early use of exchange transfusions [Sheehy et al. 1980; Costa et al. 2006]. If exchange transfusion (ET) is unsuccessful, liver transplantation may be the only lifesaving alternative. The presence of cholecystitis or choledocholithiasis, acute or chronic viral hepatitis B or C, iron overload, cirrhosis or drug induced liver injury as well as the diverse hepatic manifestations of SCD may confound the clinical presentation and pose a diagnostic dilemma for the physician. Our review focuses on the presentation, diagnosis and management of sickle cell hepatopathy.
Case presentation
A 16-year-old female with SCD and asthma presented to the emergency room with a week history of fatigue, malaise, dark urine, lower back pain, scleral icterus, and jaundice. The patient was not sexually active, had no tattoos, and denied drinking alcohol or using illicit drugs. On physical examination our patient was afebrile but ill-appearing. She had scleral icterus, cutaneous jaundice, and significant RUQ abdominal tenderness. Murphy’s sign was negative. Her liver edge was palpated 7 centimeters (cm) below the right costal margin at the midclavicular line. The spleen was nonpalpable. She had no signs of advanced liver disease.
Initial laboratory evaluation revealed a total bilirubin level of 25.3 mg/dl (normal 0.2–1.2), direct bilirubin 13.1 mg/dl (0–0.5), white blood cell count 5.6 K/cmm (4–12), hemoglobin 9.2 g/dl (11.5–15.5), hematocrit 30% (34–45), platelet count 291 K/cmm (130–400), reticulocyte count 5.6% (1–2), aspartate aminotransferase (AST) 104 U/L (8–34), alanine aminotransferase (ALT) 186 U/L (6–55), gamma glutamyl transferase (GGT) 60 IU/L (9–50), alkaline phosphatase 264 U/L (40–150) and lactate dehydrogenase (LDH) 568 U/L (125–220). Serum amylase, lipase, and electrolytes were unremarkable. Upon admission the patient received intravenous hydration and empiric intravenous antibiotic. An abdominal ultrasound revealed hepatomegaly (17 cm), nonspecific diffuse gallbladder wall thickening and gallbladder sludge. It was suggestive of intrahepatic biliary duct dilatation, but revealed no stones in the common bile duct (CBD) (Figure 1). Workup for viral hepatitis, serologic screening for Epstein—Barr virus, cytomegalovirus, ceruloplasmin, and alpha-1 antitrypsin levels were normal.
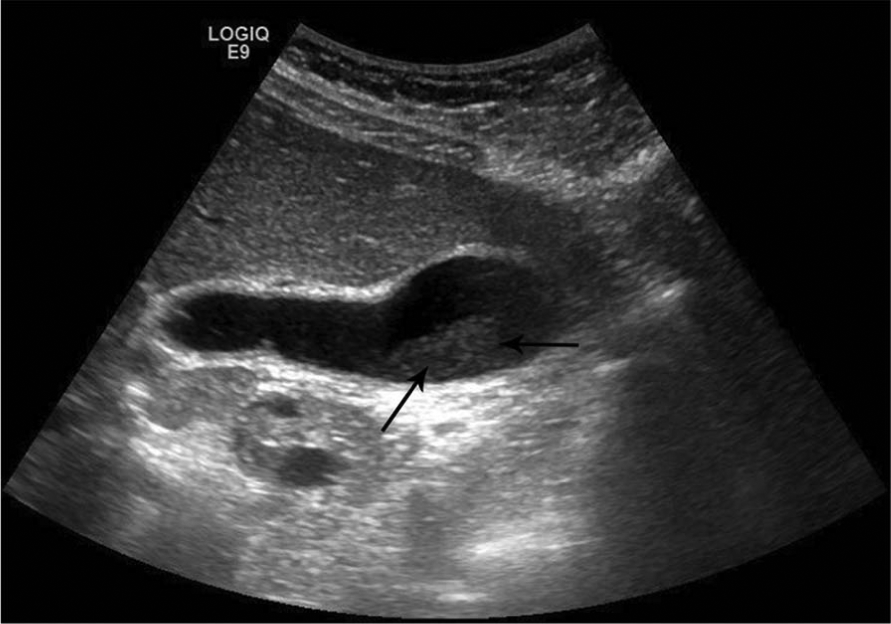
Right upper quadrant sonogram image showing nonspecific diffuse gallbladder wall thickening, gallbladder sludge (arrows).
The hospital course was complicated by coagulopathy with prothrombin time 15.7 sec (9.8–11.7), international normalized ratio (INR) 1.5 (0.8–1.2), partial thromboplastin time 30.8 sec (24.4–33.5) and acute renal failure with creatinine 1.5 mg/dl (0.6–1.1). For the persistent hyperbilirubinemia, ET with seven units of blood was performed, but the bilirubin and the other liver chemistries demonstrated only mild improvement and the RUQ pain persisted (Figure 2). The patient underwent endoscopic ultrasound (EUS) that revealed CBD sludge and gallbladder filled with small stones and sludge (Figure 3). The CBD was minimally dilated at 6.8 mm, which was not consistent with bilirubin level of 30 mg/dl. An uneventful laparoscopic cholecystectomy (LC) was subsequently performed. Over the next 24 hours there was no improvement in liver function tests (LFT). An endoscopic retrograde cholangiography was subsequently performed and the cholangiogram showed a mildly dilated CBD (7 mm) with filling defects consistent with sludge. A sphincterotomy with balloon sweep was performed and revealed large amounts of sludge and small stones (Figures 4 and 5). The patient was discharged, 72 hours later with near normal LFT.

Changes in total bilirubin, AST, ALT and ALP levels during hospitalization are plotted from admission at day 1 to discharge at day 11.

Endoscopic ultrasound image showing gallbladder filled with small stones and sludge.
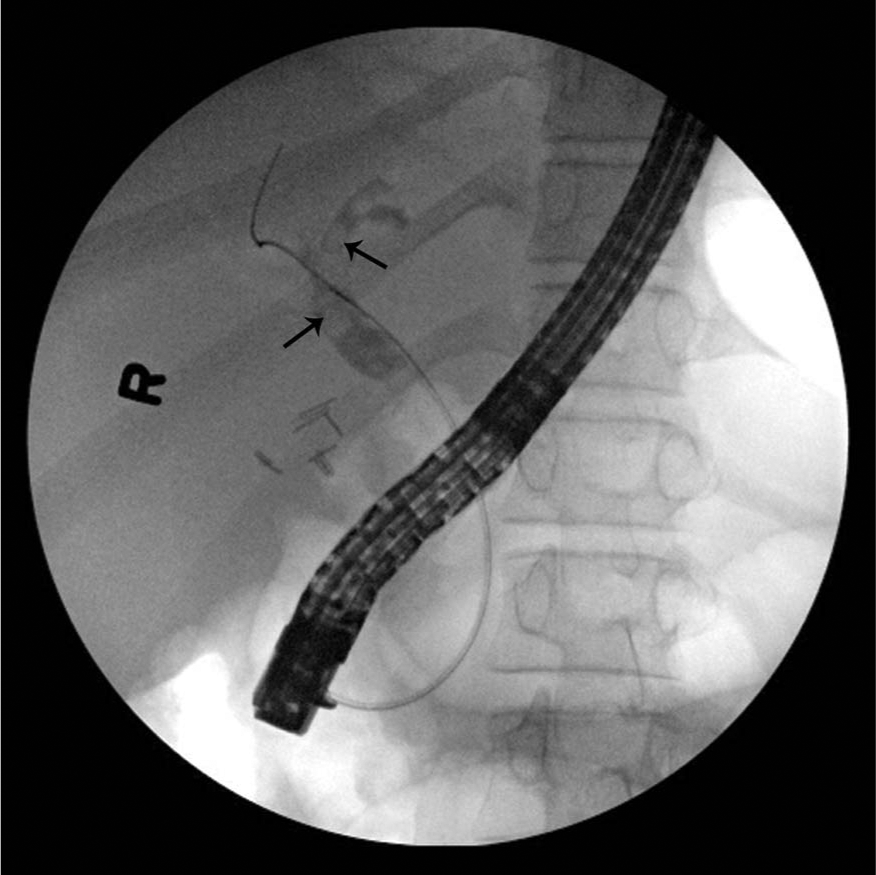
Endoscopic retrograde. cholangiopancreatography showing (arrows) dilated common bile duct and common hepatic duct with filling defect before sphincterotomy and balloon. sweep.
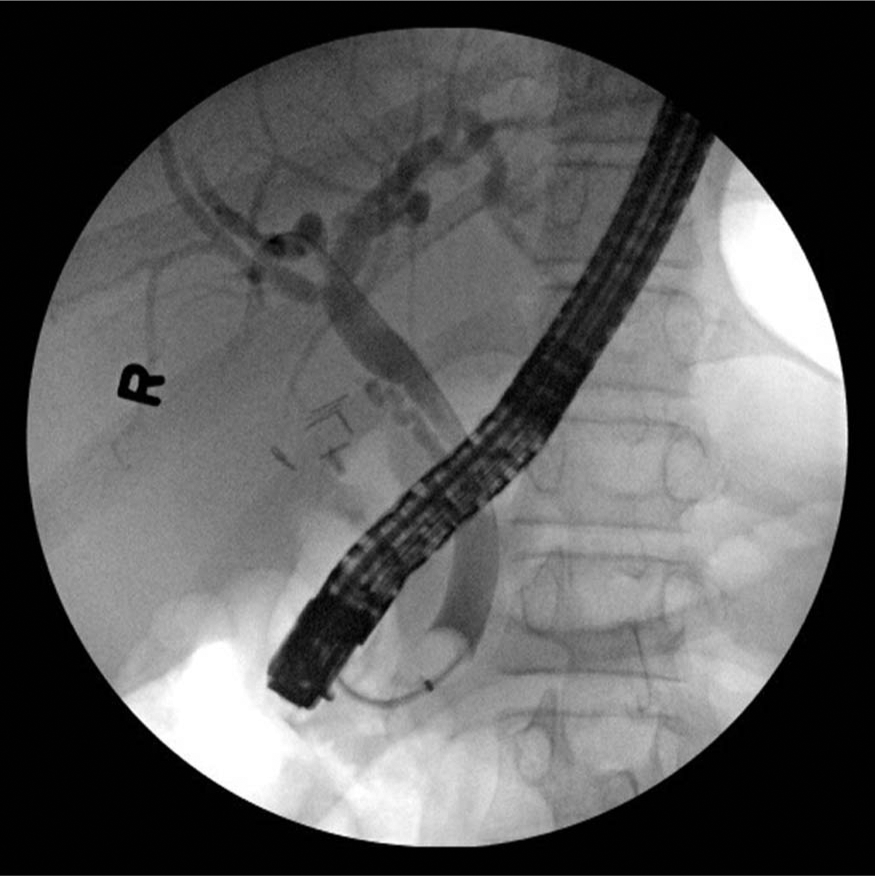
Post-sphincterotomy endoscopic retrograde cholangiopancreatography image showing resolution of filling defect shown in Figure 4.
Discussion
Liver involvement in SCD can manifest mainly as ASHC and SCIC. However, other conditions such as acute viral hepatitis, acute cholecystitis, choledocholithiasis, iron overload, advanced fibrosis or cirrhosis, and drug induced liver injury may co-exist and potentially complicate the clinical picture and delay the diagnosis and management [Song, 1957; Bauer et al. 1980].
Hyperbilirubinemia is common in patients with SCD, is mainly unconjugated and derives from chronic RBC hemolysis. Bilirubin is usually below 6 mg/dl and often is the only liver chemistry abnormality observed [Johnson et al. 1985]. Elevation of alkaline phosphatase levels may indicate cholestasis, however, bone is usually the main source [Brody et al. 1975]. In SCHC the transaminase level is usually below 300 mg/dl and total bilirubin is mainly below 15 mg/dl, primarily caused by ischemia of the hepatocytes due to sinusoidal obstruction by sickled cells. Infrequently, levels of transaminases may exceed 1000 mg/dl, which may indicate severe hepatic hypoxia. Conservative management is the usual approach and almost all cases resolve spontaneously after a couple of weeks [Diggs and Bell, 1965; Rosenblate et al. 1970; Sheehy, 1977; Schubert, 1986; Friedman, 2007]. Patients with SCIC present with remarkable clinical features such as extreme hyperbilirubinemia, with a mixed conjugated and unconjugated pattern and jaundice with hepatomegaly, coagulopathy and renal insufficiency in the absence of other acute liver disease. In severe cases, liver failure may develop. Transaminases may increase significantly (>1000 mg/dl) whereas total bilirubin level that usually ranges between 3 and 10 mg/dl in sickle cell patients may be significantly elevated (level of 273 mg/dl has been reported in literature) [Banerjee et al. 2001, Irizarry et al. 2006]. This severe hyperbilirubinemia is characteristic of SCIC and it stems from ongoing hemolysis, intrahepatic cholestasis, and renal insufficiency. It has been proposed that the pathogenesis of SCIC is associated with obstruction of the hepatic sinusoids by sickled RBC and bile with resulting vascular stasis, ballooning of hepatocytes, and local hypoxia. Moreover, SCIC has been reported to have an overall mortality rate of up to 50% in adults and 31% in pediatric patients due to coagulopathy and fulminant liver failure [Khurshid et al. 2002].
Liver biopsy may be performed to help establish the diagnosis, differentiate from other concomitant liver diseases, and determine fibrosis stage and iron deposition in Kupffer cells. However, possible complications of liver biopsy such as bleeding and injury to adjacent organs should be considered prior to such intervention, especially in the presence of coagulopathy. Liver biopsy may show sickle cell thrombi, Kupffer cell hypertrophy, dilated hepatic sinusoids, intrasinusoidal sickling of erythrocytes, centrilobular necrosis, and bile stasis. Other findings may include lymphocytic infiltration, paracentral necrosis, and bile plugs within dilated cannaliculi [Baichi et al., 2005].
Treatment of SCIC is mainly achieved with ET, close monitoring and supportive care. The ET is done in cycles, each one usually lasting a few minutes and requires that the patient’s blood be removed and replaced. Sheehy and colleagues first reported the use of ET to successfully treat SCIC [Sheehy et al. 1980]. In 2005, Ahn and colleagues studied two groups of patients based on severity of disease [Ahn et al. 2005]. Group I had maximum bilirubin level of 36.2 mg/dl and no evidence of severe hepatic dysfunction, with mortality of only 4%. Group II had a much higher level of bilirubin (maximum level of 76.8 mg/dl), prolonged prothrombin (PT) and partial thromboplastin time (PTT), and signs of encephalopathy. Mortality in group II was much higher also (64%). Among patients in group II, 7 out of 9 who received ET survived, compared to only one patient who survived among the 13 patients who did not receive ET. This result along with other reports reinforces the important role of ET in the management of SCIC [Svarch et al. 1986; O’Callaghan et al. 1995; Shao and Orringer, 1995; Stephan et al. 1995; Khurshid et al. 2002].
High hemoglobin S (HbS) levels in the setting of stress such as infection or surgery may promote sickling with consequent congestion of hepatic sinusoids due to sickle cell sequestration. Thus, the use of ET to reduce HbS levels has been shown to improve intrahepatic cholestasis due to sickle cell sequestration. Delis and colleagues described a 43-year-old patient with SCD for whom ET successfully reduced the HbS level from 35% to 7.3% and reversed the intrahepatic cholestasis that occurred after surgical resection of liver metastases due to colon cancer [Delis et al. 2006]. Similarly, in a recent review, Blinder and colleagues measured pre- and post-transplant HbS levels with a pre-transplant HbS goal of <30% and a post-transplant HbS goal of <2% in a patient with SCIC to monitor efficacy of treatment using ET [Blinder et al., 2013].
Gallstones may develop in nearly 60% of patients with SCD. These are usually calcium bilirubinate black pigmented stones [Bond et al. 1987]. About 17% have concomitant choledocholithiasis. EUS or magnetic resonance cholangiopancreatography may be useful diagnostic modalities. Acute cholecystitis may co-exist with sickle cell hepatopathy and definitive diagnosis may be facilitated by the use of ultrasound and biliary scintigraphy. Cholecystectomy is suggested in cases of SCD patients with biliary pain and gallstones; however this procedure has been associated with development of acute chest syndrome and may occasionally carry a dismal prognosis. In fact, Vichinsky and colleagues reported that acute chest syndrome occurred in 10% of patients with SCD who underwent elective surgery [Vichinsky et al. 1995]. However, operating on asymptomatic SCD patients with gallstones may be considered electively because such an approach may deter possible gallstone pancreatitis, cholecystitis, or cholangitis from occurring later in life [Hendricks-Ferguson and Nelson, 2003, Curro et al. 2007, Al-Salem and Issa, 2012]. Budd—Chiari syndrome due to thrombosis of the hepatic veins [Sty, 1982] and pyogenic abscesses due to infarction have been also reported [Lama, 1993]. Cocaine abuse by SCD patients has been associated with sickling and cocaine-induced vasospasm can precipitate hepatic crisis [Saltzman and Johnston, 1992]. Cirrhosis has been found in 29% of deceased patients with SCD, likely associated with acquisition of transfusion-related chronic hepatitis C and B and transfusion-related iron overload [Friedman, 2007].
Our patient developed coagulopathy, markedly elevated bilirubin levels and acute kidney injury typical of SCIC but she also had cholelithiasis and choledocholithiasis, which complicated the clinical picture. In the absence of advanced liver disease this patient’s hepatomegaly was likely attributed to inflammation and sinusoidal obstruction by sickled RBC. Although liver biopsy is virtually diagnostic of SCIC, in our case it was not performed. Based on the patient’s medical history, clinical presentation, laboratory findings and imaging, we hypothesized that this was a presentation of SCIC complicated by biliary obstruction. However, this diagnosis, though feasible, cannot be definitively proven in the absence of liver biopsy.
ET was performed as indicated but the RUQ pain persisted. Subsequently, the patient underwent EUS, LC and ERC with sphincterotomy that led to progressive improvement of liver chemistries. After LC there was a transient rise in the transaminase levels, most likely due to manipulation of the organ during the procedure and resection of the gallbladder from the gallbladder fossa (Figure 2). Cholecystectomy is generally recommended in symptomatic patients with SCD, however, in asymptomatic patients it remains a matter of debate [Rambo and Reines, 1986; Rutledge et al. 1986; Ware et al. 1988; Dan et al. 2009]. Emergency surgical intervention in a patient with SCD is associated with increased morbidity and mortality [Stephens and Scott, 1980; Rutledge et al. 1986], and some authors recommend early surgical intervention in asymptomatic patients [Rambo and Reines, 1986; Ware et al. 1988]. Such an approach may limit the diagnostic challenges encountered in a sickle cell patient who may present in the future with RUQ abdominal pain.
Sequential endoscopic and surgical approach of a SCD patient with suspected cholelithiasis and choledocholithiasis has been demonstrated to be safe and effective [Gholson et al. 1995]. Preoperative blood transfusions, supplemental oxygenation, and aggressive hydration pre-, peri-, and post-procedure may decrease the risk of perioperative complications [Ware et al. 1988].
Role of liver transplantation
Although liver transplantation is rare, it may be considered in patients with acute SCD hepatopathy or cirrhosis. It is associated with complications such as graft thrombosis, cerebrovascular accident, pulmonary embolism, multiple organ failure and immunosuppression-associated infections. It is associated with an overall 60% mortality in children and adults. Painful crisis episodes may continue and the graft may be affected by sickling post-transplantation [Mekeel et al. 2007; Perini et al. 2010]. Postoperative complications of liver transplantation include graft thrombosis, pulmonary embolism, stroke and multiple organ failure.
Mekeel and colleagues described a case series of three children who received liver transplants due to complications related to SCD. Two received a deceased donor graft and one a left liver graft. There were no reports of graft loss, primary non-function or vascular thrombosis. Infections, however, were common and included cytomegalovirus, bacterial pneumonia, intra-abdominal abscess, Candida albicans cholangitis and parvovirus infections, all treated successfully with antibiotics. One child who was transplanted for intrahepatic cholestasis related to SCD experienced a recurrence and subsequent graft failure. All three children suffered consequences of vaso-occlusive crises in long-term follow up (up to 5.5 years post-transplantation). The authors recommended that the hemoglobin level should be above 9 g/dl and the HbS fraction below 25% pre- and post-operatively to prevent sickling [Mekeel et al. 2007].
Adults transplanted for SCD have overall improved liver function and long-term survival. However, most of the surviving cases suffer from allograft complications (intrahepatic cholestasis, graft infarction/necrosis). Blinder and colleagues reported a case of an adult orthotopic liver transplantation for sickle cell-related liver failure, who was managed aggressively with blood transfusions in the post-operative period to maintain hemoglobin between 7 and 10 g/dl and HbS <2%, with no post-operative sickle cell related complications and no more need for blood transfusions 12 months after transplantation [Blinder et al. 2013].
Conclusion
SCIC is a rare complication of homozygous sickle cell anemia characterized by abdominal pain, hepatomegaly, extreme hyperbilirubinemia, and coagulopathy, which may lead to acute hepatic failure and death. Prompt recognition, supportive care, and early treatment with ET may result in a favorable outcome. Liver transplantation may be lifesaving in selected cases but is not curative and symptoms re-occur post-transplantation. Imaging modalities such as magnetic resonance cholangiopancreatography, computed tomography, transabdominal and EUS, and biliary scintigraphy are commonly used. Management of bile duct obstruction with endoscopic retrograde cholangiopancreatography and sphincterotomy is relatively safe and is frequently performed. However, the risks for possible pancreatitis, hemorrhage, infections, bowel perforation and anesthesia-related complications call for judicious application of this procedure. Cholecystectomy in the urgent setting may be complicated with acute chest syndrome and is not advocated by some authors; however, in our case was completely uneventful. Elective cholecystectomy in the presence of gallstones may be safely performed in asymptomatic patients with sickle cell disease. Other concomitant liver diseases including hepatitis B or C, drug-induced liver injury, hepatic vein thrombosis, liver abscesses, iron overload from blood transfusions and advanced liver disease should be also considered and ruled out in the appropriate clinical setting.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.
Informed consent
Informed consent has been obtained by the patient discussed in this case report for use of her anonymized details.
Research ethics
No ethical approval was required for this work.