
Abstract
Pancreatic neuroendocrine tumors (pNETs) differ in their clinical behavior, presentation and prognosis based on their initial histological features and disease stage. While small resectable tumors can be treated surgically, metastatic and locally advanced disease carries a significant mortality and treatment options have been limited in terms of their efficacy. Streptozocin-based regimens were the only agents available before but recent advances have improved the armamentarium to treat pNETs. Newer chemotherapeutic agents such as temozolomide, somatostatin analogs and targeted therapies including everolimus and sunitinib are now available to treat these tumors. Several combination regimens with targeted therapies and newer agents such as pazopanib are being developed and tested in ongoing trials.
Keywords
Introduction
Pancreatic neuroendocrine tumors (pNETs) account for 3–5% of pancreatic malignancies with an incidence of around 1000 cases per year in the USA [Ries et al. 2007]. However, the incidence of these tumors appears to be rising, partly due to earlier detection of asymptomatic lesions [Yao et al. 2008a]. They are a heterogeneous group of tumors with varying biology and clinical behavior based on their functionality and differentiation. Unresectable pNETs have a poor prognosis with a median survival of 24 months with distant metastatic disease [Yao et al. 2008a]. Newer agents have been developed recently to decrease the mortality related to this disease.
Most of these tumors are sporadic, although a link with hereditary syndromes including multiple endocrine neoplasia type 1 (MEN1), von Hippel–Lindau disease (vHL), neurofibromatosis type 1 and tuberous sclerosis deserves mention since it suggests the underlying pathogenesis. A study of the genetic mutations in nonfamilial pNETs has revealed mutations in MEN1, DAXX/ATRX and the mammalian target of rapamycin (mTOR) pathway, again leading to the hope of being able to stratify patients in the future to tailored therapy [Jiao et al. 2011]. Further literature has supported the importance of the mTOR pathway [Missiaglia et al. 2010]. Elevated levels of vascular endothelial growth factor (VEGF) also led to a hypothesis of the efficacy of VEGF inhibitors in pNETs [Zhang et al. 2007].
The neuroendocrine tumors (NETs) are classified by the World Health Organization based on their differentiation in order to assess their biological behavior and their potential for a malignant phenotype. Low grade (grade 1) and intermediate grade (grade 2) tumors have a solid, glandular pattern, with fairly uniform nuclei, and finely granular cytoplasm but are separated based on the proliferative index. Grade 3 tumors are poorly differentiated and are considered to have a high malignant potential and are further classified as small cell or large cell neuroendocrine carcinomas. The mitotic count and the Ki-67 index are part of the classification as detailed in Table 1.
World Health Organization 2010 classification.

The well differentiated pNETs can be further classified based on their functionality and are divided into functioning and nonfunctioning tumors. The functioning tumors include insulinomas, gastrinomas, glucagonomas, VIPomas and somatostatinomas. The nonfunctioning tumors are more frequent and account for almost 60% of these tumors.
Management of pancreatic neuroendocrine tumors
Management of pNETs is based on a multidisciplinary approach involving assessment by a surgeon for resectability, evaluation for and management by an endocrinologist of any active peptide production and systemic treatment by an experienced oncologist in cases of unresectable disease. Also playing a major role in pNETs with hepatic disease are radiation oncologists for radiofrequency ablation (RFA) of solitary liver metastases and interventional radiologists for transarterial chemoembolization in the case of multiple hepatic metastases.
Surgery is the treatment of choice for locoregional nonfunctioning tumors that are resectable with enucleation. Larger resectable lesions can be treated with distal pancreatectomy (for distal lesions) or pancreatoduodenectomy (for lesions in the head of the pancreas) with lymph node resection. For functioning tumors including insulinomas, glucagonomas and VIPomas, stabilization of glucose levels, use of diazoxide, octreotide and correction of electrolyte abnormalities are required before proceeding to surgery in resectable cases. Gastrinomas require use of proton pump inhibitors and sometimes octreotide preoperatively. However, locoregional unresectable disease or metastatic disease management has been a challenge but has been a field undergoing dynamic changes in the past few years.
Hepatic radiofrequency ablation and transarterial chemoembolization
Hepatic RFA is an option for small hepatic metastatic lesions that are not amenable to resection in cases of predominant disease in the liver and has been shown to provide prolonged symptomatic relief in reported case studies [Gillams et al. 2005; Mazzaglia et al. 2007].
Transhepatic arterial embolization with or without chemotherapy is useful in patients with unresectable hepatic predominant disease who are not candidates for RFA due to the size of the lesions. The possible side effects of fever, pain and transaminitis need to be evaluated before deciding on this procedure, although a case report has shown a median response duration of 17 months in patients with carcinoids [Gupta et al. 2003].
Chemotherapy
Several chemotherapies have been investigated to treat unresectable pNETs. Streptozocin-based regimens including streptozocin–doxorubicin and streptozocin–5-fluorouracil (5-FU) have shown efficacy in the past but come with the burden of toxicity and cumbersome regimens. Newer regimens have been evaluated in trials and these include temozolomide, capecitabine and oxaliplatin alone or in combination with targeted agents. Temozolomide regimens in combination with thalidomide, everolimus or bevacizumab have shown a response rate of 25–45% [Chan et al. 2012; Kulke et al. 2010]. Although temozolomide has shown some efficacy in retrospective studies [Ekeblad et al. 2007], combination with capecitabine in a recent study showed a significant radiological response of 70% with an overall progression-free survival of 18 months [Strosberg et al. 2011]. Oxaliplatin has shown efficacy in combination with capecitabine and other agents, including bevacizumab, in phase II trials [Bajetta et al. 2007; Venook et al. 2008]. However, the wide array of chemotherapy options still give limited disease-free survival and there were previously few options available for the treatment of pNETs that have progressed on chemotherapeutic agents (Table 2).
Chemotherapeutic agents in pancreatic neuroendocrine tumors.

mOS, median overall survival; mPFS, median progression-free survival; NA, not available; ORR, overall response rate.
Hormonal treatment
Somatostatin analogs have long been used to control symptomatic disease, including VIPomas and glucagonomas. The PROMID trial showed an increased time to tumor progression when octreotide long acting release (LAR) was used in small bowel carcinoid tumors (14.3 versus 6 months) [Rinke et al. 2009]. Ongoing trials including the CLARINET trial will evaluate the role of this class of drugs in pNETs in combination with other agents and will help to define their role in asymptomatic patients.
Immunotherapy
Interferon α has been used historically for the treatment of advanced disease, mostly in patients who have symptomatic and progressive disease on somatostatin analogs. Its use is limited because of significant side effects and because of the lack of clear effect on tumor progression. The use of interferon α is likely to decline further given the advent of new targeted therapies [Arnold et al. 2005].
Targeted therapy
The field for treatment options for unresectable or metastatic pNETs changed dramatically in 2011 when two new targeted agents, sunitinib and everolimus, were approved by the US Food and Drug Administration (FDA) for this indication.
Sunitinib
Sunitinib is a multi-tyrosine kinase inhibitor which has shown to have antitumor and antiangiogenic properties. It inhibits VEGF receptors (VEGFR-1, 2 and 3), platelet-derived growth factor receptor (PDGFR-A and B), KIT and colony stimulating factor 1 receptor (CSF-1R).
In a RIP1-Tag2 transgenic mouse model of pancreatic islet-cell tumors, sunitinib was found to have efficacy by reducing endothelial cell density and pericyte coverage of tumor vessels, leading to reduction in tumor size and an increased survival of the mice [Pietras and Hanahan, 2005]. Based on a significant response seen in preclinical studies, a phase I study conducted with sunitinib for solid tumors including NETs showed evidence of disease response with a dose of 50 mg given daily for 4 weeks out of 6 weeks [Faivre et al. 2006]. This led to a multicenter phase II trial in which 106 patients which included 66 patients with pNETs were treated with the same dose schedule and showed an overall objective response rate of 16.7% and 68% had stable disease (SD) [Kulke et al. 2008]. The overall response rate (ORR) was 16.7%, the median time to progression was 7.7 months and the 1-year survival rate was 81.1%.
A randomized, double-blind, placebo-controlled phase III trial compared the tyrosine kinase inhibitor sunitinib at a dose of 37.5 mg daily with placebo and showed a definite progression-free survival of 11.4 months in contrast to just 5.5 months for placebo [p < 0.001, hazard ratio (HR) 0.42) [Raymond et al. 2011a]. A total of 171 patients with well differentiated pNETs were included in this multinational trial. The overall survival (OS) was 30.5 compared with 24.4 months, which did not reach clinical significance, possibly due to treatment crossover [Raymond et al. 2011b]. The ORR was 9.3%, suggesting disease stabilization as the cause of the longer progression-free survival. The most frequent adverse events (AEs) in the sunitinib group were diarrhea, nausea, vomiting, asthenia and fatigue.
Hematological side effects including anemia and thrombocytopenia can occur but grade 3/4 severity is seen only in 5–10% of patients receiving sunitinib. Toxicities seen with this tyrosine kinase inhibitor that need to be monitored for include the risk of hepatotoxicity, reduction in left ventricle ejection fraction (LVEF), QT prolongation and adrenal insufficiency, which could manifest with stress responses.
Serial monitoring of a patient on sunitinib includes a complete blood count (CBC), liver function tests, chemistries, urinalysis for proteinuria, an electrocardiogram and an echocardiogram to evaluate the LVEF at baseline and periodically if patients have cardiac risk factors. Drug interactions need to be evaluated carefully before initiating treatment or any change in therapy (Table 3).
Drug interactions of sunitinib.

CYP3A4, cytochrome P450 3A4; VEGF, vascular endothelial growth factor.
Management recommendations for monitoring and dose titration for side effects are summarized in Table 4.
Sunitinib side effects and their management.

ADLs, activities of daily living; ALT, alanine aminotransferase; AST, aspartate aminotransferase; LLN, lower limit of normal; LVEF, left ventricle ejection fraction; WNL, within normal limits.
Everolimus
Everolimus (RAD-001; Afinitor, Novartis Pharmaceuticals, Basel, Switzerland) has been demonstrated to be effective in a variety of cancers, including advanced renal cell carcinoma and in progressive hormone receptor positive, human epidermal growth factor receptor 2 negative breast cancer, in conjunction with aromatase inhibitors.
Everolimus is a 40-O-(2-hydroxyethyl) derivative of sirolimus. By binding to and blocking FKBP-12, everolimus inhibits mTOR, an intracellular serine/threonine kinase which signals downstream transduction of receptor tyrosine kinases, resulting in blocking of cell growth, cellular proliferation and angiogenesis. Inhibition of mTOR has a significant antiproliferative effect on pNET cell lines [Missiaglia et al. 2010]. Studies have demonstrated that the loss of the neurofibromatosis (NF1) gene, which regulates the activity of mTOR, leads to mTOR activation and has been shown to be associated with the pathogenesis of carcinoid tumors in the mediastinum, ampulla of Vater and the duodenum [Johannessen et al. 2005; Tan et al. 1996; van Basten et al. 1994]. Mutations in the tuberous sclerosis complex (TSC)1/2 gene, which is an endogenous mTOR inhibitor normally expressed in neuroendocrine cells, are associated with the development of islet cell tumors [Eledrisi et al. 2002; Plank et al. 1999; Verhoef et al. 1999]. Activation and expression of insulin-like growth factor 1 (IGF-1), which stimulates the mTOR signaling pathway, is also associated with the proliferation of pNET cells, while inhibition of mTOR suppresses NET growth [O’Reilly et al. 2006]. These findings have led to the development of everolimus as a therapy for advanced pNETs.
The first study of everolimus in pNETs was a single-arm, single-institution phase II trial that evaluated everolimus plus octreotide LAR in 60 patients with either carcinoid (n = 30) or islet cell tumors (n = 30) and found a partial response (PR) rate of 22%, SD rate of 70% and a progressive disease (PD) rate of 8%, with overall median progression-free survival (mPFS) of 60 weeks. ORR and mPFS were higher in the 30 patients with pNETs. These results compared favorably with chemotherapy and targeted agents. The antitumor activity of everolimus was thought to be independent of octreotide, which has a ORR of only 2–3% on its own [Yao et al. 2008b].
The RAD001 in Advanced Neuroendocrine Tumors (RADIANT) trials, the largest studies conducted to date in advanced NETs, were designed to assess the efficacy of everolimus in NETs of different origins. The RADIANT-1 trial, an open-label, nonrandomized, international phase II trial, confirmed the promising antitumor findings of everolimus in progressive pNETs. The trial evaluated 160 patients who received either everolimus alone (n = 115) or in combination with octreotide LAR (n = 45). In the everolimus-alone arm, there were 11 PRs (9.6%), 78 with SD (67.8%), 16 with PD (13.9%) and mPFS of 9.7 months. By comparison, in the everolimus/octreotide arm, there were 2 PRs (4.4%), 36 patients with SD (80%), no patients with PD (0%) and mPFS of 16.7 months [Yao et al. 2010]. The RADIANT-2 trial evaluated everolimus/octreotide LAR versus placebo/octreotide LAR in extrapancreatic NETs and demonstrated significantly improved PFS and significantly reduced risk of disease progression with the addition of everolimus [Pavel et al. 2011].
The landmark RADIANT-3 trial, an international prospective, randomized, phase III study, evaluated everolimus as first-line treatment for advanced pNETs. The study evaluated 410 participants with progressive, advanced, low-grade or intermediate-grade pNETs and compared everolimus (n = 207) with placebo (n = 203). The mPFS in the everolimus arm was 11.0 versus 4.6 months in the placebo arm (HR 0.35), a 65% reduction in the estimated risk of progression or death. The proportion of patients alive and progression free at 18 months was 34% with everolimus compared with 9% with placebo. Although no significant difference in OS was found, 73% of the placebo patients crossed over to the everolimus arm, confounding any survival benefit. The ORR of everolimus was relatively low (5%), indicating that much of the benefit was from minor tumor shrinkage and disease stabilization [Yao et al. 2011].
The RADIANT trials demonstrated that everolimus is generally well tolerated in patients with pNETs. The AEs were mostly grade 1 and 2 and long-term daily administration was well tolerated and generally manageable [Pavel et al. 2011; Yao et al. 2008b, 2010, 2011]. The most common drug-related AEs were stomatitis or aphthous ulcerations (64%), rash (49%), diarrhea (34%), fatigue (31%) and infections (23%). The most common grade 3 or 4 AEs were stomatitis (7%), anemia (6%) and hyperglycemia (5%). Everolimus was associated with thrombocytopenia (4%), mild lymphopenia and neutropenia. There was a 23% incidence of drug-related infections, with the majority grade 1 and 2 with 2% grade 3 or 4. Pneumonitis (12%; 2% grade 3 or 4) and interstitial lung disease (2%), both associated with sirolimus derivatives, were important clinical concerns but were effectively manageable. Everolimus in combination with octreotide was associated with similar adverse effects and was determined to be well tolerated (Tables 5 and 6) [Pavel et al. 2011; Yao et al. 2008b].
Everolimus adverse effects and their management.

ADLs, activities of daily living.
Everolimus drug interactions.

CYP3A4, cytochrome P450 3A4
Multiagent Therapy with Everolimus
The rationale for combining octreotide and everolimus is that upregulation of the upstream IGF pathway is thought to be a resistance mechanism for everolimus and octreotide reduces serum IGF-1 levels in patients with solid tumors. Everolimus in combination with octreotide LAR has been shown to be efficacious and well tolerated and is indicated in patients with functional pNETs who require somatostatin analog therapy.
Temozolomide in combination with everolimus is being evaluated in a phase I–II study and has shown promising initial results. Of 17 patients, 6 had a PR (35%), 9 had SD (53%) and 2 had PD (12%). Accrual has completed and an updated analysis is anticipated [Kulke et al. 2010] [ClinicalTrials.gov identifier: NCT00576680].
Bevacizumab combined with everolimus is also under study. In an assessment of the effect of bevacizumab on tumor blood flow utilizing a functional computed tomography scan, bevacizumab decreased tumor blood flow and the addition of everolimus to bevacizumab resulted in a further decrease in tumor blood flow. Furthermore, bevacizumab combined with everolimus demonstrated antitumor activity in low to intermediate grade NETs. Preliminary results of a multicenter phase II trial show that bevacizumab with another mTOR inhibitor, temsirolimus, resulted in a marked improvement in response rate compared with the response rates of everolimus monotherapy in the RADIANT trials. Confirmed PR was seen in 11 of the first 25 (44%) evaluable patients and 20 of 25 (80%) patients progression free at 6 months. A major CALGB trial evaluating everolimus and octreotide with or without bevacizumab in patients with metastatic pNETs has completed accrual with an expected primary completion date of December 2014 [ClinicalTrials.gov identifier: NCT01229943].
Several other combined approaches are presently being evaluated, including mTOR and IGF-1R inhibition, mTOR inhibition plus pan-analog of somatostatin receptors (SOM230), mTOR and epidermal growth factor receptor inhibition, and dual mTORC1 and mTORC2 and dual phosphatidylinositol 3 kinase and mTORC1/2 inhibition.
At present, there are two FDA-approved targeted agents based on the above trials (Table 7). Combination treatment options involving a chemotherapeutic agent and a biological agent or an immunomodulatory drug have been evaluated with the hypothesis of improving response and survival rates in various phase II trials (Table 8).
US Food and Drug Administration approved targeted therapy.
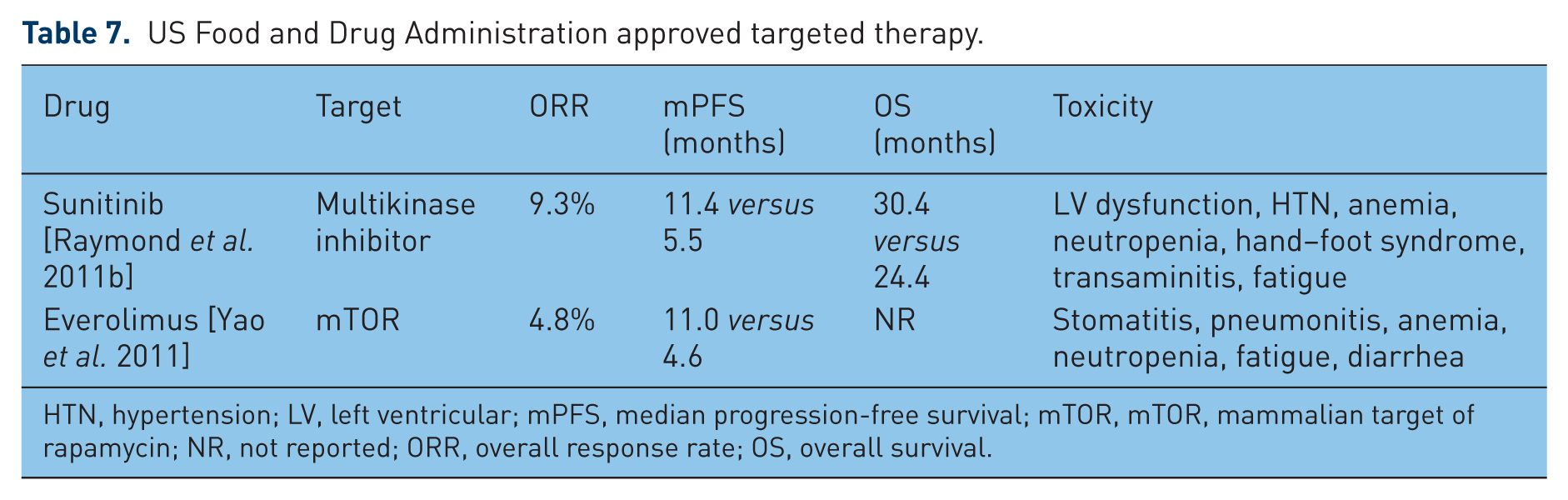
HTN, hypertension; LV, left ventricular; mPFS, median progression-free survival; mTOR, mTOR, mammalian target of rapamycin; NR, not reported; ORR, overall response rate; OS, overall survival.
Combination therapy for pancreatic neuroendocrine tumors.

mPFS, median progression-free survival; NR, not reported; ORR, overall response rate; OS, overall survival.
Biomarkers
Biomarkers may be used to assist in both the diagnosis and post-treatment follow up in patients with pNETs.
Established biomarkers
For well differentiated NETs, when sufficient tumor material is available for histologic review, immunohistochemistry and other ancillary techniques may not be required for diagnosis. In less well differentiated or poorly differentiated tumors, specific markers that may be used to establish neuroendocrine differentiation include chromoganin A (CgA), synaptophysin and cytokeratin. Ki-67 (MB-1) and the mitotic rate are markers of tumor cell proliferation that have been proposed as grading criteria for NETs and have been shown to correlate with outcome [Rindi et al. 2007]. In a phase II study of 60 patients, including 7 patients who consented to a core needle biopsy before and after treatment, tumor Ki-67 was used as a biomarker for tumor proliferation, which was decreased following everolimus treatment [Yao et al. 2008b].
Blood markers are also used to diagnose pNETs and evaluate response to therapy. These include CgA, pancreatic polypeptide and specific serum hormone levels in functional pNETs.
Chromogranin A
CgA is the most commonly secreted and measured hormone associated with all types of gastroenteropancreatic NETs. Plasma levels are thought to correlate with tumor burden. Levels are elevated in 60–70% of patients with pNETs, including 75% of functioning pNETs [Modlin et al. 2010; Panzuto et al. 2004]. Increased CgA in patients with NETs has been associated with a poor PFS and survival [Janson et al. 1997; Jensen et al. 2007]. CgA can be followed as a tumor marker if elevated at baseline. Of the patients with pNETs in the RADIANT-1 trial, a CgA response was seen in 50.7% of patients who received everolimus, and an early CgA response was associated with a significantly prolonged PFS [Yao et al.].
Chromogranins B and C are less sensitive indicators of gastroenteropancreatic NETs compared with CgA and they are not used clinically [Verhoef et al. 1999].
Pancreatic polypeptide
Pancreatic polypeptide is another nonspecific biochemical marker for nonfunctioning pNETs, but it has a relatively low sensitivity (63%) and specificity (81%) when used alone. Compared with CgA alone, the combined use of pancreatic polypeptide plus CgA improved sensitivity for nonfunctioning pNETs from 68% to 93% [Panzuto et al. 2004].
Serum hormone levels in functioning pancreatic neuroendocrine tumors
For patients with functioning pNETs, the levels of the secreted hormone represent a more specific tumor marker. For gastrinomas, serum gastrin levels may be followed; for insulinomas, serum proinsulin, insulin/glucose ratio and C-peptide levels; for VIPomas, serum VIP levels; for glucagonomas, serum glucagon, glucose and CBCs; for other functioning pancreactic NETs, serum somatostatin, calcitonin and parathyroid hormone related peptide levels may be followed as per National Comprehensive Cancer Network (NCCN) guidelines.
Novel biomarkers
Neuron-specific enolase
In the RADIANT-1 trial, 30–50% of patients had an elevated neuron-specific enolase (NSE), with 68–73% showing a response to everolimus. Further evaluation of NSE as a biomarker is however needed before it can be used in clinical practice [Yao et al. 2008b].
Circulating tumor cells
Khan and colleagues investigated the role of circulating tumor cells (CTCs) as a biomarker in NETs, including a sample of pNETs (31 of 138; 22%) [Khan et al. 2012]. The authors enrolled patients with advanced NETs who were commencing systemic or local therapy. They found that patients with one or more CTCs detected by the Cell Search (Veridex LLC, Raritan, NJ, USA) technology were more likely to have worse PFS and OS. In multivariate analysis the HR for survival in patients with one or more CTCs was 5.1 (95% confidence interval 1.5–7.3). In addition they compared samples measured prior to beginning therapy and 3–5 weeks after starting therapy and found that an increase of more than 33% in the number of CTCs was a strong predictor of poor response to therapy with worse PFS and OS.
Placental growth factor
Fischer and colleagues evaluated placental growth factor (PlGF) as a prognostic biomarker in NETs et al. [Fischer et al. 2012]. PlGF is a homolog of VEGF, which is recognized to be overexpressed in NETs. The VEGF pathway is an important target for drug therapy in pNETs, with both the approved agent sunitinib (a multi-targeted tyrosine kinase inhibitor) and experimental therapies with bevacizumab (a monoclonal antibody against the VEGF pathway). The authors measured PlGF levels in serum and in tumor samples via immunohistochemistry. They found that PlGF levels were elevated in pNET samples and serum compared with control pancreatic tissue and control serum. Prospective investigation validated this finding of elevated serum PlGF levels in both pNETs and NETs. The authors also reported that there was a suggestion that elevated PlGF levels are correlated with shorter time to progression (at least in NETs) and that this warrants further prospective evaluation as a predictive biomarker (Table 9).
Details of the abstracts presented at the 2012 American Society of Clinical Oncology annual meeting addressing the role of established and novel biomarkers on predictive and prognostic parameters of pancreatic neuroendocrine tumors.

5-HIAA, hydroxyindoleacetic acid; CgA, chromogranin A; CI, confidence interval; CTC, circulating tumor cell; HR, hazard ratio; NET, neuroendocrine tumor; OS, overall survival; PFS, progression-free survival; PlGF, placental growth factor; pNET, pancreatic neuroendocrine tumor; RCT, randomized controlled trial.
The future of pancreatic neuroendocrine tumor therapy
Several promising agents are being evaluated as single agents or in combination with known agents (Table 10). Temsirolimus, another mTOR inhibitor, has been evaluated in a phase II trial for advanced NETs, including carcinoids and pNETs, with an ORR of 5.6% and a median time to progression of 6 months [Duran et al. 2006]. Temsirolimus is being investigated in combination with bevacizumab to assess for an improvement in response rate and the final trial results are awaited [Hobday et al. 2012].
Promising agents for treatment of pancreatic neuroendocrine tumors.

CR, complete response; IGF-1R, insulin-like growth factor 1 receptor; LAR, long acting release; mPFS, median progression-free survival; mTOR, mammalian target of rapamycin; ORR, overall response rate; PDGFR, platelet-derived growth factor receptor; pNET, pancreatic neuroendocrine tumor; PR, partial response; RR, response rate; SD, stable disease; VEGFR, vascular endothelial growth factor receptor.
Another potential target is the IGF pathway since both the ligand (IGF) and the cognate receptor (IGF-1R) are frequently expressed in NETs. The use of a monoclonal antibody that blocks IGF-1R (AMG 479) is being evaluated in a phase II trial but the initial results have not shown evidence of a disease response [Kulke et al. 2012].
Several tyrosine kinase inhibitors are being investigated for the treatment of pNETs. Sorafenib is a tyrosine kinase inhibitor which targets the VEGF-R2 and PDGFR-B tyrosine kinase domains. VEGF and PDGF as well as their receptors have been shown to be overexpressed in pNETs and tumor growth and angiogenesis are thought to be stimulated by activation of these pathways. In a phase II trial, sorafenib showed a PR of 10% [Hobday et al. 2007].
Pazopanib is a multikinase inhibitor of VEGF-R1, VEGF-R2, VEGF-R3, PDGF-R and c-KIT-R. A multicenter single-arm nonrandomized phase II trial looked at the efficacy of this agent in well and moderately differentiated pNETs and carcinoids. An overall clinical benefit rate (CR + PR + SD) of 85.7% was seen at 6 months [Enrique Grande, 2012]. Another study of pazopanib and LAR showed an ORR of 17% with a median survival of 11.7 months [Phan et al. 2012].

Algorithm suggestion for treatment of metastatic pancreatic neuroendocrine tumors.
Histone deacetylase inhibitors are another class of drugs that are on the horizon to be evaluated for the treatment of pNETs. Romidepsin was administered in a phase I trial and has shown evidence of disease stabilization [Doss et al. 2008].
Discussion
NETs consist of a diverse group of tumors composed of cells showing neuroendocrine cell differentiation (secretory granules), a subset of which can be further classified by their dominant secretory products. Although it was thought that the neuroendocrine cells that give rise to NETs migrated from the neural crest to the gut endoderm, it is now apparent that enteropancreatic neuroendocrine cells originate from multipotent stem cells that give rise to all epithelial cell types in the gastrointestinal tract and pancreas. NETs show heterogeneity in morphologic, functional and clinical features. Although pNETs represent a small percentage of all pancreatic tumors (1.3%), the prevalence of these tumors is significant (9.9% of all pancreatic tumors) and the incidence is increasing. In tertiary oncology centers, the majority of patients with malignant pNETs present with advanced stage tumors and approximately 65% of patients present with unresectable or metastatic disease.
Prior to 2011, the only approved agent for unresectable disease was streptozocin, which was approved prior to 1984 after demonstrating some efficacy in early trials (either alone or in combination with doxorubicin or 5-FU). Further studies have questioned the efficacy of streptozocin and there had not been any new drugs approved in the last 20 years. As a result, patients with unresectable pNETs had a poor prognosis. The median survival time for patients with distant metastatic disease is 24 months; the 5-year survival rate of patients with metastatic disease is 30–40% and this has not changed for 20 years. The effort to find improved therapeutics for pNETs was bolstered by the observation that there are several inherited cancer syndromes which are associated with pNETs (including MEN1, vHL, NF1 and TSC) [Jensen et al. 2008]. Unfortunately the underlying genetic abnormalities in these syndromes are relevant in only a subset of the sporadic pNETs. A global gene expression analysis of pNETs revealed that at least two important genes in the mTOR pathway (TSC2 and PTEN) were downregulated in 85% of primary tumors [Missiaglia et al. 2010]. In addition, aberrant expression of several tyrosine kinase receptors and overexpression of VEGF have been noted in pNETs [Zhang et al. 2007].
The NCCN guidelines recommend observation with imaging and marker assessment every 3–12 months in people who have asymptomatic, stable and unresectable pNETs. For symptomatic well to moderately differentiated unresectable disease, sunitinib and everolimus have become the first-line treatment options in addition to chemotherapeutic agents known from before. Chemotherapy drug regimens available include capecitabine, dacarbazine, doxorubicin, 5-FU, streptozocin and temozolomide, which have been used with varying response rates. Octreotide treatment, as discussed above, is also an option if other agents cannot be used. Hepatic therapies, including cytoreductive or ablative agents, are available, although the evidence for these therapeutics is still developing.
Is there an optimal therapy?
The side-effect profile, drug efficacy and cost-effectiveness analysis can help guide decisions regarding treatment options in patients with pNETs. Yao recently discussed patient selection criteria in a plenary session based on the recent trials (oral communication, European Society of Medical Oncology, Plenary Session, 2012). There is no head-to-head comparison between everolimus and sunitinib and validation of predictive biomarkers for selection of one agent over the other remains arbitrary. However, patient characteristics, including a history of uncontrolled hypertension and heart disease, would make sunitinib a less favorable option given the side effects of reduction in ejection fraction and increased blood pressure seen as a possible side effect.
A total of 24% of the 410 patients in the RADIANT-3 study had gastrinoma, glucagonoma, VIPoma, insulinoma or somatostatinoma, implying that everolimus may be used across the spectrum of pNET types. Across subgroups, the trial found that the benefit of everolimus was maintained across subgroups of age, sex, race, geographic region, performance status and receipt of prior therapy (chemotherapy, radiation or octreotide).
However, sunitinib would be a better option in people with underlying severe lung disease given the small but significant risk of pneumonitis seen with everolimus therapy. Table 11 summarizes the above considerations for review when deciding on targeted therapy for pNETs.
Considerations for review when deciding on targeted therapy for pancreatic neuroendocrine tumors.

Cost-effectiveness analysis of sunitinib and everolimus
A cost-effectiveness analysis comparing everolimus and sunitinib in a simulated cohort of patients with advanced progressive pNETs has yielded interesting data in favor of everolimus [Casciano et al. 2012].
The analysis estimated the total and incremental life years (LYs) and quality-adjusted life years (QALYs). The incremental cost-effectiveness ratios (ICERs, cost/QALY) were calculated as the cost per LY gained and cost per QALY gained. In the final analysis, everolimus was found to be more cost effective. It was associated with an incremental 0.448 LY gained (0.304 QALY) at an incremental cost of US$12,673, resulting in an ICER of US$28,281/LY gained (US$41,702/QALY gained). The ICER was comparable to other known oncology drugs currently in use.
Conclusion
Chemotherapy remains the treatment of choice in poorly differentiated locally advanced tumors or tumors with symptoms related to rapid disease progression. Sandostatin is an excellent option in functional tumors for symptom control. Both sunitinib and everolimus have brought significant benefits for patients with pNETs and can be used in well to moderately differentiated metastatic pNETs. The results of phase III studies underline important progress in the management of patients with pNETs and should be considered as a valuable addendum to the current treatment options. One criticism of the results of both phase III studies would be that significant improvement in PFS is just a surrogate marker because significant improvement in OS has not been shown. However, the design of a phase III study with OS as the primary endpoint and a study power of 90% would require an estimated sample size of 2800 patients to show that a survival benefit of 4 months is significant. Looking at the low incidence rate of pNETs, successful recruitment of a sufficient number of subjects into such a study would be very unlikely. The oral form of administration of both drugs offers a great convenience to patients in an outpatient setting. The benefit of these two agents can be maintained across various subgroups, including functional/nonfunctional, chemonaive/treated patients. Although both drugs produce AEs, these AEs are generally manageable with dose reduction, temporary interruption of therapy or both. It is clinically relevant to choose patients for specific drugs based on the typical drug-related AEs of each drug. Additional predictive biomarkers of response to sunitinib and everolimus for selection of patients with pNETs are required to help gauge disease response at an early stage. The question of whether continuous or intermittent treatment or alternating these agents could be an option also remains unanswered at present and will require evaluation.
Trials with promising agents targeting different pathways are eagerly awaited to shape the future for the treatment of pNETs. Combination therapies involving chemotherapy agents and targeted agents will hopefully improve the response rates and the PFS in this otherwise relatively chemotherapy-resistant disease (Table 12).
Take home message.

NET, neuroendocrine tumor; pNET, pancreatic neuroendocrine tumor.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
Dr Saif is on the speaker bureau for sunitinib.