
Abstract
Cardiomyopathy is a heart muscle disorder that cannot be attributed to ischemic heart disease or abnormal loading conditions such as hypertension and valvular heart disease. It can lead to the development of arrhythmia and heart failure, and is also the leading cause of sudden cardiac death in young people. Early detection is vital for delaying or preventing complications. This review discusses the four major subtypes, i.e. hypertrophic, dilated, restricted, and arrhythmogenic cardiomyopathy, which are likely to be encountered in primary care.
Clinical case scenario
A 47-year-old presented to her GP with a 6-month history of reduced exercise tolerance, chest pain, dyspnoea and peripheral oedema. Her medical history included frequent palpitations and one episode of syncope following intense exercise in her 30s. Investigations at the time, including a 24-hour electrocardiogram (ECG) and echocardiography (ECHO), were normal. The family history included a grandparent who died suddenly in his 30s. Positive findings on examination included an ejection systolic murmur and peripheral oedema. A subsequent ECG revealed left ventricular hypertrophy (LVH) and deep q-waves in the anterolateral leads. The patient was referred urgently to cardiology. An ECHO showed asymmetric hypertrophy of the anterolateral left ventricular wall measuring 22 mm, mild mitral regurgitation and an ejection fraction of 56%. There was no evidence of outflow tract obstruction. She was diagnosed with hypertrophic cardiomyopathy and referred to the inherited cardiac conditions clinic. Genetic testing was negative. Further evaluation with cardio-pulmonary exercise testing and cardiac magnetic resonance imaging (CMR) was normal. Her risk of sudden cardiac death was estimated to be 0.7% per annum, which is low. She was started on bisoprolol and eplerenone and allowed to continue with moderate exercise.
Hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a heterogeneous genetic disorder that causes asymmetric hypertrophy of the left ventricular wall. Inheritance is usually autosomal dominant, although rare autosomal recessive and x-linked forms exist. The prevalence of HCM is estimated to be around 0.2–0.5% in the general adult population. It affects all ethnicities and genders equally and is the leading cause of sudden cardiac death (SCD) in young athletes (Maron and Maron, 2013).
Aetiology
The aetiology is due to one or more gene mutations that code for sarcomere components, the cardiac myocyte's contractile unit. The sarcomere comprises thick and thin filaments that move across each other to cause cardiac muscle contraction. HCM develops due to mutations that prolong contraction and delay sarcomere relaxation (Iavarone et al., 2022). Many genes have been implicated, but actual evidence for causality is available only for a few (Table 1). Around 60% of HCM cases can be accounted for by known gene mutations. However, the presence of a known mutation does not always result in the development of HCM, indicating that other factors are involved. The variable expression and penetrance of the disease also appear to be age dependent, i.e. approximately 50% of patients begin to show phenotypic changes on an echocardiogram by adolescence to early 20s. This rises to around 75% when these individuals reach their 50s (Charron et al., 1997).
HCM causative genes.
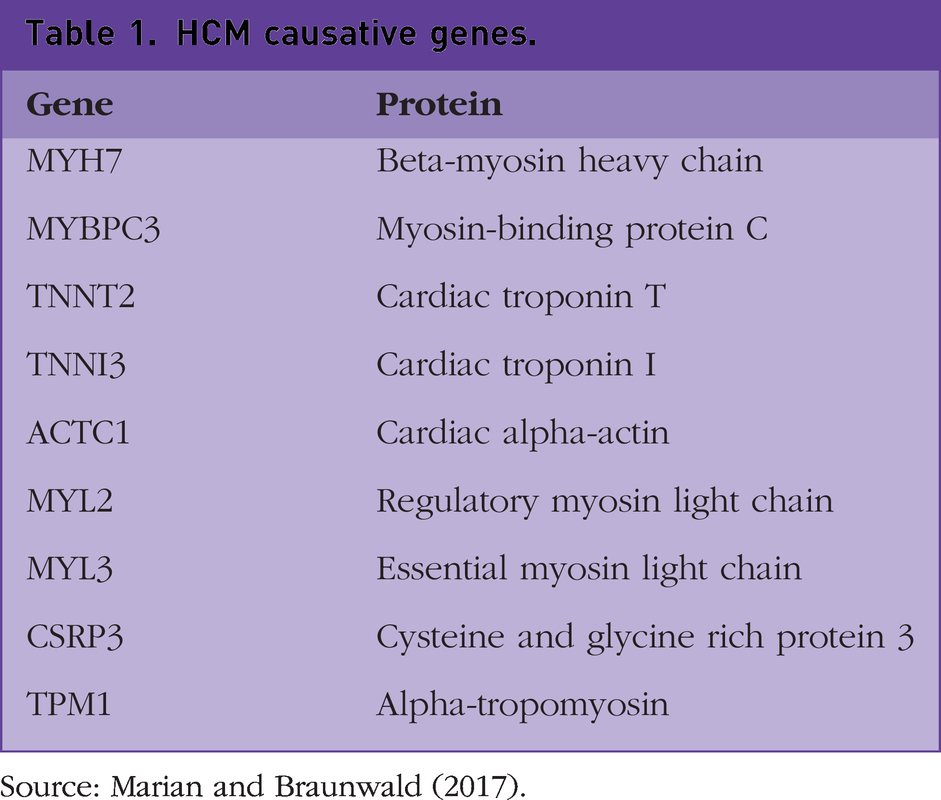
Source: Marian and Braunwald (2017).
History and clinical features
Most individuals are initially asymptomatic, and symptoms emerge as a sign of disease progression. Presentation is variable and may include dyspnoea, reduced exercise tolerance, fatigue, palpitations, dizziness, dyspnoea, chest pain, exertional pre-syncope/syncope, peripheral oedema, acute heart failure, cardiac arrest and sudden death (Ciarambino et al., 2021). Family history can be the key to the diagnosis, and enquiries should be made for up to three generations regarding sudden unexplained death, heart failure or stroke at an early age, i.e. at 35 or less. Examination findings may include an ejection systolic murmur, which may be intensified by the Valsalva manoeuvre and reduced by squatting. If there is associated mitral regurgitation (MR), a holosystolic murmur, which radiates to the axilla, may be heard. Other examination findings may include displaced/double apex beat and signs of heart failure (Ciarambino et al., 2021).
Investigations
Investigations should include a 12-lead ECG, ECHO and Holter monitoring to detect ventricular arrhythmia. The ECG can be normal in about 10% of cases. In the remainder, characteristic findings include voltage criterion for LVH, non-specific ST and T wave changes and deep dagger-like q waves in the anterolateral (L1, AVL, V5-6) and inferior leads (Leads II, III and AVF). An example ECG is shown in Fig. 1.
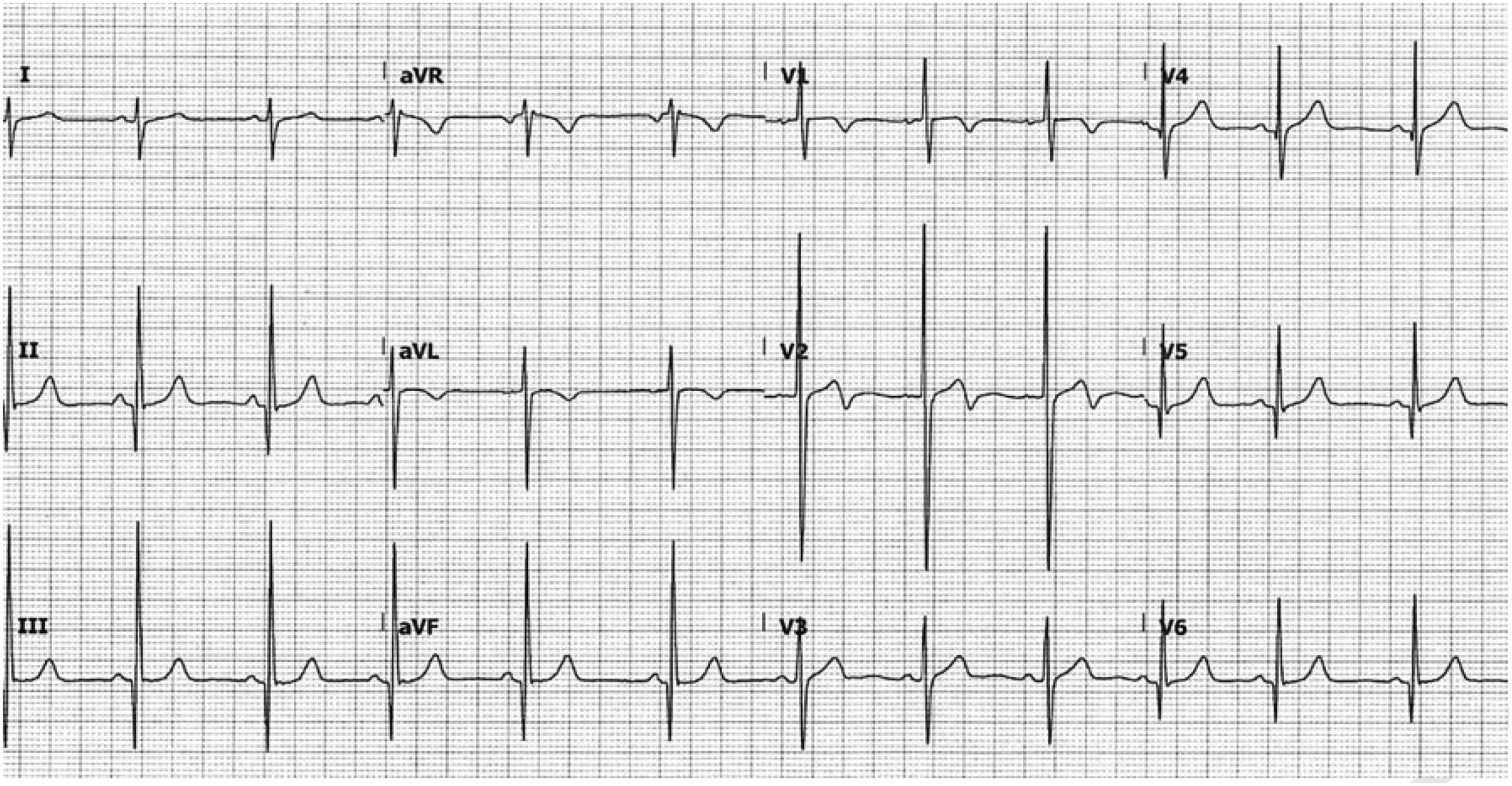
Characteristic findings on ECG in patients with HCM.
An echocardiogram is diagnostic if it shows asymmetric left ventricular wall hypertrophy in diastole greater than 15 mm in a non-dilated ventricle. In first-degree relatives, the threshold is lower at 13 mm for screening and early diagnosis (Gartzonikas et al., 2022). The ECHO may show left ventricular outflow tract obstruction (LVOTO), systolic anterior motion (SAM) of the mitral valve, MR and left atrial dilation. LVOTO is seen in about two-thirds of patients and can be present at rest or be provoked by exertion. The movement of the mitral valve leaflet towards the ventricular septum during systole (SAM) can also contribute to obstruction.
A 24-hour Holter monitor is also essential, as around 20–30% of patients have runs of non-sustained ventricular tachycardia (VT), a risk factor for sudden cardiac death (Iavarone et al., 2022). Further tests are indicated for risk stratification, e.g. evaluation of LVOTO with exercise testing and CMR for detecting myocardial fibrosis (Sebastian et al., 2023). If a diagnosis of HCM is made, genetic testing is indicated to facilitate family screening, as relatives who do not have HCM-associated mutations can be re-assured (Ommen et al., 2020). Without an identifiable genetic cause, screening of all first-degree family members should be continued 1–3 yearly in children and every 3–5 years in adults. (Ommen et al., 2020).
Management
Asymptomatic individuals diagnosed with HCM require follow-up every 1–2 years (Gatzonikas et al., 2022). Co-morbidities such as hypertension and hypercholesterolemia should be managed according to local or national guidelines. All patients should be assessed for the risk of SCD, which can be estimated using symptoms, family history, 48-hour ambulatory ECG, exercise stress testing and echocardiography findings (European Society of Cardiology, 2014). SCD is largely preventable with an implantable cardioverter defibrillator (ICD) (Maron et al., 2022). A history of prior cardiac arrest or evidence of sustained VT warrants ICD insertion. For the remainder, the presence of any of the following, including recent unexplained syncope, family history of SCD at a young age, recurrent non-sustained VT, left ventricular hypertrophy > 30 mm, hypotension associated with stress testing or significant cardiac fibrosis seen by cardiac MRI, should be considered for an ICD (Ommen et al., 2020). Patients at low risk of SCD do not require prophylactic ICD insertion.
Symptomatic patients should be initiated on beta-blockers, e.g. bisoprolol, metoprolol or propranolol. If intolerant to beta-blockers, L-type calcium channel blockers, e.g. verapamil or diltiazem, can be used. In HCM with LVOTO, Disopyramide can be added second line if symptoms persist. If fluid overload symptoms occur, loop, thiazide diuretics, or aldosterone antagonists may be needed. The development of heart failure with a reduced ejection fraction (< 50%) should be managed according to national guidelines (National Institute for Health and Care Excellence (NICE), 2018). In LVOTO, dehydration and excess alcohol should be avoided, as these can reduce ventricular pre-load, exacerbating LVOTO. Strenuous and intense exercise, e.g. sprinting, weight lifting, and competitive sports, should be prohibited, although moderate exercise can be continued. Illicit drugs such as cocaine and amphetamines must be strictly avoided (Sebastian et al., 2023). Patients with LVOTO who remain symptomatic despite optimal pharmacotherapy should be referred for consideration of myomectomy or septal ablation (Ommen et al., 2020).
Arrhythmia is common, with up to 24% of individuals developing atrial fibrillation (AF) (Siontis et al., 2014). In HCM, AF increases the risk of thromboembolic stroke (around 3.75% per annum), regardless of CHA2DS2VASC score and progression to end-stage heart failure; thus, all patients should be considered for anticoagulation (Guttmann et al., 2014). Patients should also be referred to an arrhythmia clinic for rhythm restoration, which may be achieved pharmacologically or by catheter ablation. In a small proportion of patients, usually with non-obstructive HCM, there is progression to end-stage heart failure despite optimal medical therapy. These individuals should be considered for a heart transplant.
Dilated cardiomyopathy
Dilated cardiomyopathy (DCM) is the most common form of cardiomyopathy. The estimated prevalence is 1 in 250 individuals (Schultheiss et al., 2019). DCM is characterised by a dilated and poorly contracting left or both ventricles, with a reduced ejection fraction (less than 45%), that cannot be explained by ischemic heart disease or conditions that increase cardiac load such as hypertension and valvular heart disease (McNally and Mestroni, 2017). DCM can develop at any age, including very young children. It is more common in males and individuals of African descent. DCM may progress to heart failure and is associated with an increased risk of arrhythmia and SCD (Jefferies and Towbin, 2010). In children, where it is commonly idiopathic, it is a devastating disease leading to end-stage heart failure within 1–5 years of diagnosis, requiring heart transplantation (Nakatani, 2009).
Aetiology
DCM is not a single disease entity, but rather a collection of disorders culminating in a structurally dilated heart with poor contractility. It has many causes (Table 2), including genetic, acquired, mixed, secondary to systemic disease or idiopathic (Jefferies and Towbin, 2010). Genetic causes are the most common, accounting for up to 48% of cases, with more than 50 genetic mutations implicated (McNally and Mestroni, 2017). Inheritance is mainly autosomal dominant. Mutations in TITIN (TTN), the largest protein in the body and an important component of the sarcomere unit, account for about 25% of familial cases (Ciarambino et al., 2021).
Causes of DCM.

Source: Hanselmann et al. (2020) and Japp et al. (2016).
Infections are the next most common cause, mainly due to viruses, accounting for around 30% of cases. Detection of viral genomes, by PCR amplification of DNA from myocardial tissue, is useful for directing therapy with antivirals (Jefferies and Towbin, 2010). DCM may have an autoimmune aetiology and can also develop secondary to systemic autoimmune disorders, including systemic lupus erythematosus, rheumatoid arthritis and systemic sclerosis (Schultheiss et al., 2019). Chronic alcohol consumption, defined as intake greater than 80 g per day for more than 5 years, is a well-established cause of DCM, accounting for around 10% of cases in men and 3% in women (Piano and Phillips, 2014). Long-term cocaine use is also associated with the development of DCM. Doxorubicin, an anthracycline drug, is commonly used to treat many paediatric cancers, including acute leukaemia, with a high degree of success. However, cardiac complications, including DCM, occur in up to 60% (Schultheiss et al., 2019).
A rare but important cause of DCM is a pregnancy-related condition known as peripartum cardiomyopathy (PCM). Patients present with symptoms of heart failure in late pregnancy or within 5 months post-partum. The condition is relatively rare, with an estimated prevalence of 1:1000 to 1:10 000 (Ersboll et al., 2017). The cause of PCM is unknown, but may have a genetic basis, including mutations in the TTN gene (Rodriguez Ziccardi and Siddique, 2020). Risk factors for PCM include age >30, African ethnicity, multiparity, Development or history of pre-eclampsia/eclampsia, cocaine and use of tocolytics to delay delivery (Ciarambino et al., 2021).
History and clinical features
In addition to general medical history and examination, enquiry should be made regarding sudden unexplained death, heart failure or stroke at a younger age, e.g. at 35 or less, in at least three generations (Mestroni et al., 1999). Substance abuse should also be enquired about, as well as travel history; the latter is relevant for identifying infectious causes e.g. Lyme disease and Chagas disease (Japp et al., 2016). The clinical features include reduced exercise tolerance, dyspnoea, peripheral oedema, persistent fatigue, palpitations, dizziness and chest pain, acute heart failure and sudden cardiac death. The examination may reveal cachexia, a gallop rhythm, a holosystolic murmur that radiates to the axilla consistent with MR, peripheral oedema and signs of acute heart failure.
Investigations
Routine investigations should include blood tests for B-type natriuretic protein (BNP) or N-terminal brain natriuretic protein (NT-BNP), which, if elevated, indicate heart failure. The ECG may be normal or show non-specific changes, e.g. enlarged p wave, AV nodal block, bundle branch block and lateral T wave inversion. Premature ventricular beats are common. ECHO findings include a spherical heart with dilated atria and ventricles. MR is common. Doppler studies show increased end-diastolic volume, systolic dysfunction and an ejection fraction of less than 45%.
Genetic testing should be arranged if the history suggests a familial cause. If testing is positive, screening is recommended for all first-generation family members. Mutation carriers, if asymptomatic, should be screened with an ECG and ECHO every 1–3 years until the age of 60. Family members who do not carry the mutation can be reassured and discharged (Hanselmann et al., 2020; Japp et al., 2016). Family screening is also recommended for idiopathic cases where the phenotype includes cardiac conduction defects, e.g. AV node blocks (Japp et al., 2016).
Further tests in secondary care may include angiography to exclude ischemic causes, Holter monitoring to detect arrhythmias, exercise testing, CMR and endomyocardial biopsy. CMR is the gold standard for assessing functional impairment and the extent of fibrosis, a risk factor for SCD. Endomyocardial biopsy is indicated in all patients with suspected inflammatory or infectious causes as this can reveal the underlying aetiology and direct treatment decisions (see below).
Management
Treatment is as per guidelines for heart failure with reduced ejection fraction, e.g. angiotensin-converting enzyme inhibitors (ACEi)/angiotensin receptor blockers, beta-blockers, aldosterone antagonists, ACEi/neprilysin inhibitor combination, nitrates and hydralazine (NICE, 2018). About one-third of DCM patients respond well to medical therapy, indicating a good prognosis. Immunosuppressive therapy may be beneficial in virus-negative DCM (confirmed by biopsy). If viral genomes are detected, antiviral treatment with interferon-beta is beneficial (Kuhl et al., 2003). Patients not responding to medical therapy require referral for cardiac resynchronisation or pacemaker/ICD insertion. AF is a common complication, and patients should be referred for rhythm control and may require long-term anticoagulation. In end-stage heart failure, patients should be considered for a heart transplant.
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCM) is a rare form of cardiomyopathy, so its prevalence has been difficult to estimate. It can affect all ages and appears to be slightly more common in women. The prognosis is correlated with the age of diagnosis, with worse outcomes if identified at a younger age (Muchtar et al., 2017). RCM is characterised by stiffened, non-compliant ventricles, resulting in reduced filling and stroke volume. The underlying problem is severe diastolic dysfunction. Systolic function is usually preserved until the latter stages of the disease (Gowda et al., 2022).
Aetiology
RCM is mainly an acquired disease, with cardiac amyloidosis being the most common cause. Other acquired causes include sarcoidosis and haemochromatosis. Primary familial causes are less common compared to the other types of cardiomyopathies. In the latter, inheritance is usually autosomal dominant. There is overlap with gene mutations implicated in HCM and DCM, i.e. mutations that result in sarcomere dysfunction (Rai et al., 2009). Interestingly, HCM can initially present with restrictive ventricular filling before the hypertrophic phenotype becomes evident (Rai et al., 2009). The causes of RCM have been classified according to histological types (Table 3).
Causes of RCM.
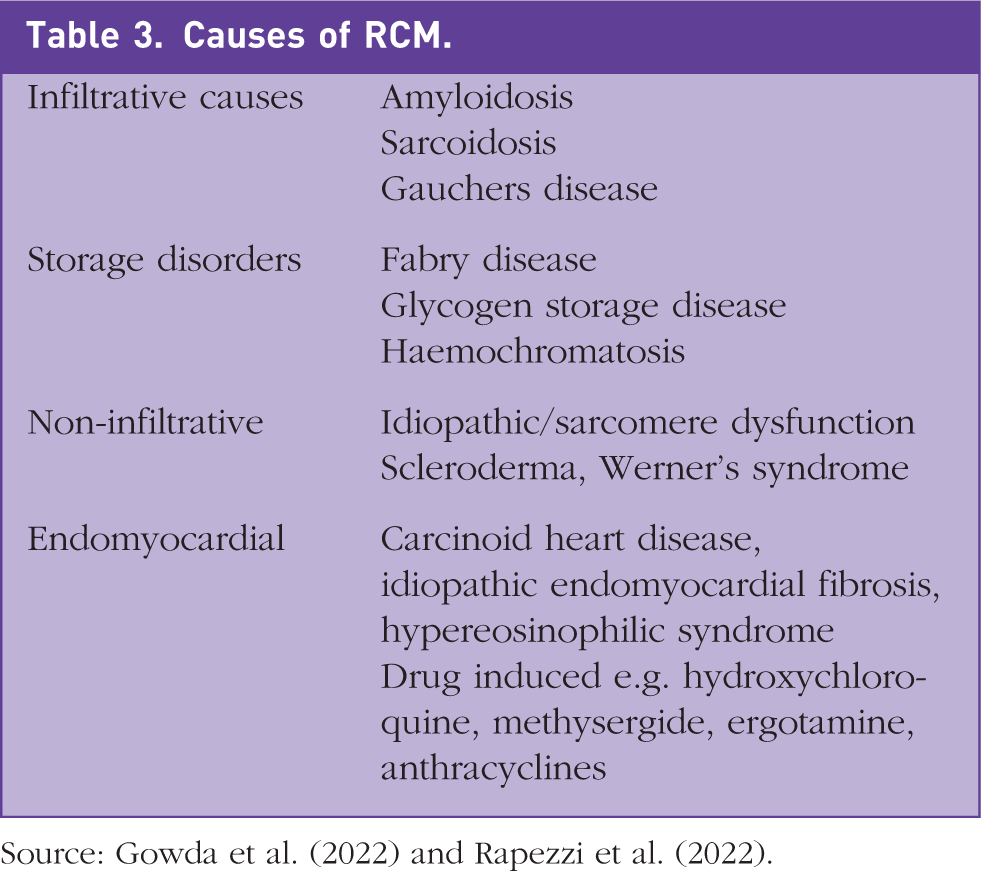
Source: Gowda et al. (2022) and Rapezzi et al. (2022).
History and clinical features
Symptoms include palpitations, reduced exercise intolerance, chest pain, dyspnoea, exertional syncope, peripheral oedema, orthopnoea and pulmonary oedema. Examination findings may include an elevated jugular venous pressure, peripheral oedema, hepatomegaly and ascites (right-sided heart failure signs) and left-sided/congestive heart failure signs, e.g. dyspnoea, orthopnoea, paroxysmal nocturnal dyspnoea, pulmonary oedema. AF is common due to atrial enlargement. Depending on the underlying cause, there may be other clinical clues pointing to the diagnosis, e.g. skin pigmentation and development of diabetes in haemochromatosis and cutaneous manifestations of sarcoidosis.
Investigations
ECG findings are non-specific and include enlarged or bifid p waves, short PR interval, pre-excitation, AV (atrioventricular) block, RBBB (right bundle branch block) and low QRS voltages (Rapezzi et al., 2022). ECHO findings that suggest RCM include bi-atrial enlargement, marked tricuspid regurgitation, and reduced ventricular filling due to diastolic dysfunction. Ventricular wall thickness is generally normal, but can be increased in some infiltrative and storage disorders (Table 3). Cardiac catheterisation shows elevated end-diastolic ventricular pressures (Gowda et al., 2022). CMR can help determine the cause of RCM, e.g. late gadolinium enhancement in the subendocardial region is highly suggestive of amyloidosis. Endomyocardial biopsy is reserved for cases where non-invasive tests have failed to identify a cause.
Management
Treatment depends on the cause. Idiopathic and Familial causes have a poor prognosis, with a median survival of less than 5 years and 1.4 years in adults and children, respectively (Gowda et al., 2022). These cases need an early referral to heart failure services to consider a heart transplant. Symptomatic treatment involves using diuretics for fluid overload, e.g. loop diuretics or aldosterone antagonists, but with care to avoid hypovolemia, which may exacerbate symptoms by further reducing stroke volume and cardiac output. Physiological compensation may include an elevated heart rate to increase cardiac output and vasoconstriction to improve blood pressure (Muchtar et al., 2017). Thus, standard heart failure regimens, including ACEi and beta-blockers, may not be well tolerated. AF is common and leads to worsening symptoms. In these patients, restoring sinus rhythm is more important than rate control and requires urgent referral to an arrhythmia clinic. Anti-coagulation is also indicated regardless of the CHA2DS2VASC score (Rapezzi et al., 2022). Disease-modifying treatment may benefit other causes of RCM, e.g. prevention of iron overload, immunosuppression and stem cell transplantation (Rapezzi et al., 2022).
Arrhythmogenic cardiomyopathy
Arrhythmogenic cardiomyopathy (AC) is more commonly known as arrhythmogenic right ventricular cardiomyopathy (ARVC), due to initial findings suggesting that the disease was limited to the right ventricle. However, both ventricles are often involved, prompting the new terminology. AC is a rare cause of cardiomyopathy. The prevalence is estimated to be between 1:1000 and 1:5000 (Corrado et al., 2017). It is more common in men, and typically presents in adolescence to early adulthood. It is the second most common cause of sudden death in young athletes after HCM (Corrado et al., 2017).
Aetiology
AC is usually inherited in an autosomal dominant manner. In most cases, the genetic fault results in abnormal cardiomyocyte adhesion, which leads to cell death and fibro-fatty replacement of the affected myocardial tissue. Fibrotic tissue interspersed with functional cardiomyocytes causes abnormal conduction and a high risk of developing dangerous arrhythmias, including VT and AF (Corrado et al., 2017).
History and clinical features
In addition to general medical history and examination, a family history should be obtained explicitly enquiring about sudden death at a young age, i.e. 35 or less. Patients are often asymptomatic and come to the fore following sudden unexplained syncope or cardiac arrest. AC can progress to heart failure and mimic DCM.
Investigations
The diagnosis requires fulfilling several major and minor criteria set out by the task force for diagnosing AVRC (Corrado et al., 2021). The criteria include detection of known mutations, family history, characteristic ECG changes, the presence of ventricular arrhythmias, ECHO and CMR findings. An endomyocardial biopsy may also be required. An epsilon wave in lead V1 on the ECG is a hallmark of AC and is seen in about 50% of cases (Fig. 2).
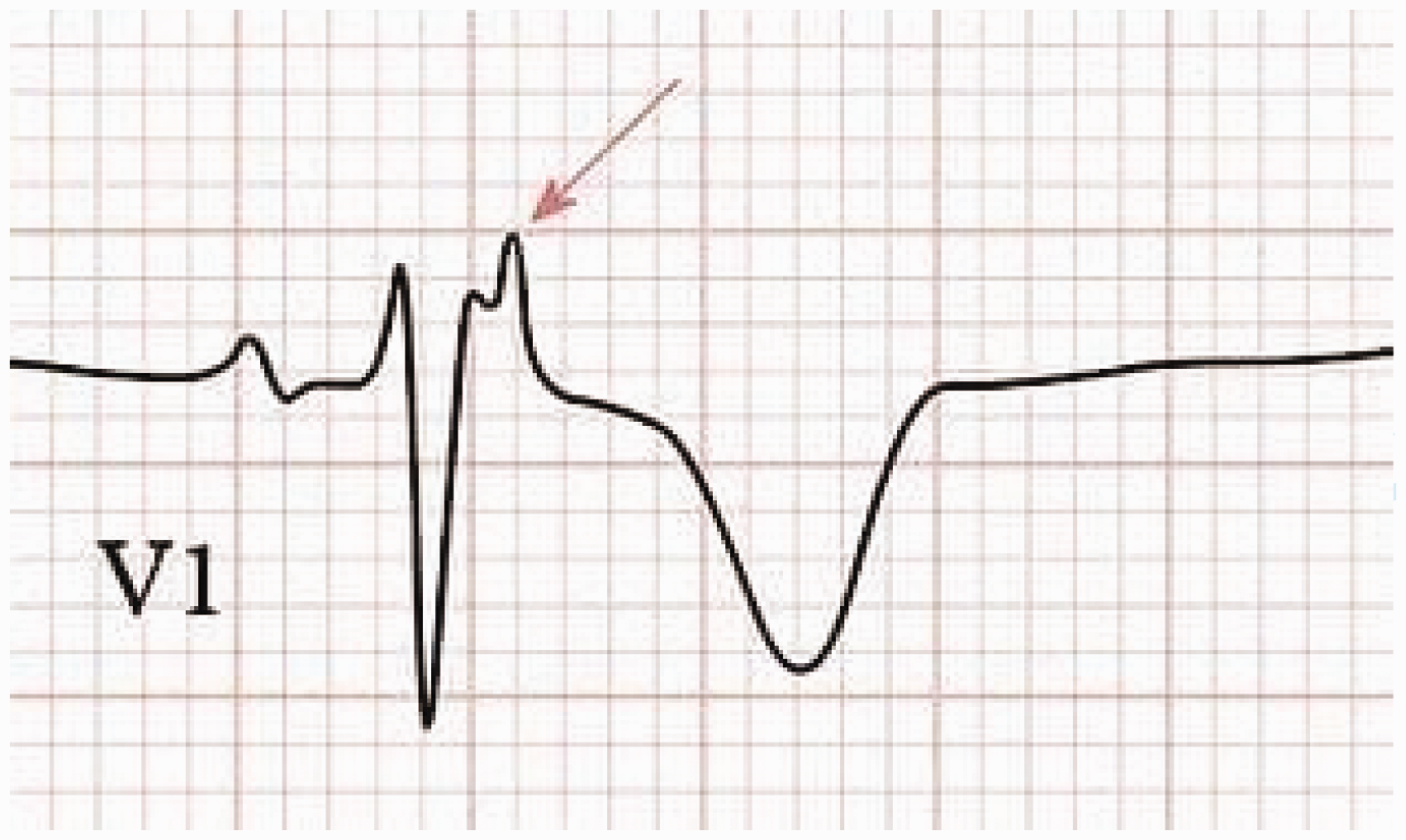
The epsilon wave in ARVC (identified by the arrow).
Management
All patients with suspected AC require urgent referral to an inherited cardiac conditions clinic. The condition is associated with a high risk of sudden cardiac death;, therefore, urgent assessment for prophylactic ICD insertion is indicated. High-risk features include prior cardiac arrest, non-sustained VT and severe systolic dysfunction. Risk scores of > 10% per year indicate an urgent need for an ICD. Patients with scores of 1–10%, which may be due to a history of exercise-induced syncope, non-sustained VT or moderate systolic dysfunction, are considered at intermediate risk and should be considered for an ICD. If the risk is estimated to be <1%, an ICD is not recommended (Corrado et al., 2017).
Lifestyle measures are also necessary, e.g. prohibiting strenuous/competitive exercise, as this is associated with an increased risk of a life-threatening arrhythmia. Medical therapy with beta-blockers has been shown to reduce the risk of ventricular arrhythmia. The development of AF is common and anti-coagulation should be considered (Corrado et al., 2017).
Conclusion
Cardiomyopathy is a common heart disorder likely to be encountered in general practice. Although the diagnosis and treatment are the remits of specialists, GPs have a role to play in early recognition, initial investigations and referral. GPs are also likely to be involved in the long-term care of these patients and need to be aware of relevant management guidelines and potential complications, such as the development of AF.
Key points
Cardiomyopathy is the leading cause of heart failure and sudden death in young patients Family history is a crucial part of the workup and should include at least three generations, enquiring about stroke, heart failure, or sudden cardiac death aged 35 or less AF in the context of HCM requires anti-coagulation regardless of the CHA2DS2VASC score due to the increased risk of stroke DCM can present at any age, including very young children and should be considered in all patients presenting with unexplained cardiac symptoms RCM in children requires early referral to a transplant team AC is associated with a high risk of ventricular arrhythmias and requires urgent referral for an ICD insertion