
Abstract
Background:
The objective of this review is to provide a practical update on endpoint selection for noninferiority (NI) studies in percutaneous coronary intervention studies.
Methods:
A PubMed search was conducted for predefined terms to explore the use of NI designs and intrapatient comparisons to determine their current importance. Sample size calculations for the most frequently used endpoints with NI hypotheses were done to increase statistical awareness.
Results:
Reported NI trials, with the most frequently chosen clinical endpoint of major adverse cardiac events (MACE), had NI margins ranging from 1.66% to 5.00%, resulting in patient populations of 400–1500 per treatment group. Clinical study endpoints comprising of MACE complemented with rates of bleeding complications and stent thrombosis (ST) are suggested to conduct a statistically and clinically meaningful NI trial. Study designs with surrogate endpoints amenable to intrapatient randomizations, are a very attractive option to reduce the number of necessary patients by about half. Comparative clinical endpoint studies with MACE and ST/bleeding rates to study a shortened dual antiplatelet therapy (DAPT) in coronary stent trials are feasible, whereas ST as the sole primary endpoint is not useful.
Conclusions:
Expanded composite clinical endpoints (MACE complemented by ST and bleeding rates and intrapatient randomization for selected surrogate endpoints) may be suitable tools to meet future needs in device approval, recertification and reimbursement.
Introduction
Clinical studies are essential to demonstrate the safety and efficacy of drugs or medical devices. In these studies, the primary endpoints are the measures of success by which treatment groups can be compared. Based on these primary endpoints, the size of the study population will be calculated. To design a meaningful study with clinically relevant endpoints, properly powered with a sufficient number of patients, and with the potential to impact guideline recommendations can be a cumbersome task. Even experienced trialist are not protected from designing a trial that may be inclusive despite its large study population. Very recently, a large randomized controlled trial (RCT) with a patient recruitment of over 3000 patients was reported to use an incorrect noninferiority (NI) margin, thereby impacting the main conclusion of the trial. 1 (SORT OUT IX trial, ClinicalTrials.gov identifier: NCT02623140). This gives us reason to believe that there is a paucity of advice for the clinical scientist planning NI trial designs.
Furthermore, given the upcoming European Medical Device Regulation (EU MDR, article 61), 2 which will be effective in May 2020, a stronger focus on clinical evidence with proprietary data for medical devices will be enforced for all device manufacturers. Therefore, a comparison of clinical safety, efficacy and usability between new and predicate devices, or existing and competitor devices, based on well-designed clinical studies appear to be essential to gain or maintain regulatory access to the European market.
Owing to the fact that device-related event rates are typically in the single-digit percentage range, NI study designs have been popular. Byrne and Kastrati commented that large NI margins may be statistically sound but may not represent the clinical scenario. 3 However, their call for increased resources to properly study and compare devices may be limited by small differences in event rates leading to very large patient populations. These, in turn, would require logistics that would facilitate rapid patient inclusion to avoid long recruitment windows, which may affect data quality or may even introduce bias into the data set.
Based on a previous review, 4 the objective of this collaborative work is to provide guidance on sample size calculations for NI studies in the field of percutaneous coronary angioplasty. In particular, we will elaborate on the following topics which seem to be of current interest.
Recommendations for acceptable NI margins in angiographic and clinical endpoint trials.
Sample size calculations for the most prominent endpoints such as major adverse cardiac events (MACE) and late lumen loss (LLL) with commonly accepted NI margins.
Statistical methods to reduce sample sizes or to use existing data sets.
Methods
Literature searches were conducted on PubMed/Medline and EMBASE with prespecified search protocols. Inputs for sample size calculations were obtained from recently published studies investigating drug-eluting stents (DES) or drug-coated balloons (DCB).
Sample size calculations
Study designs for clinical endpoints rely on binomial proportions and their difference between two independent groups. The corresponding test hypotheses are as follows 4 :
Ho: The event rate π1 in the treatment group is higher than, or equal to, the event rate π2 in the control group plus a NI margin Δ.
Ha: Event rate π1 in the treatment group is lower than π2 in the control group plus a NI margin Δ.
The basic principle of the sample size calculation is that the probability of a positive test for a given study sample (i.e. power) is greater if the study sample is larger. In fact, there is a mathematical relationship between expected binomial proportions π1 and π2, the NI margin Δ, the significance level alpha, the number of patients n, and the power. So when π1, π2, and Δ are known, fixing alpha to 5% and the power to, for example, 80%, we can calculate the minimum sample size n.
Likewise, test hypotheses for angiographic endpoints such as LLL can be formulated 4 :
Ho: LLL in the treatment group is higher than, or equal to, LLL in the control group plus a NI margin ΔLLL.
Ha: LLL in treatment group is lower than LLL in the control group plus a NI margin ΔLLL.
Assuming normally distributed variables, the number of patients are estimated based on the significance level alpha, desired power, standard deviations (SD), and the NI limit. For all sample size calculations, nQuery Advisor version 7.0 (Cork, Ireland) was used.
Literature search
The search terms listed in Table 1 were used to explore current interests as outlined in the Introduction.
Search terms for endpoints in coronary interventions with a focus on coronary stents (until August 1, 2019).

Results
The prespecified literature searches (search #1) were conducted and can be summarized in Figure 1. Literature searches #2 and #3 revealed a total of two references. The results of the literature searches can be divided in NI for clinical endpoints (Table 2) and NI for angiographic endpoints (Table 3).
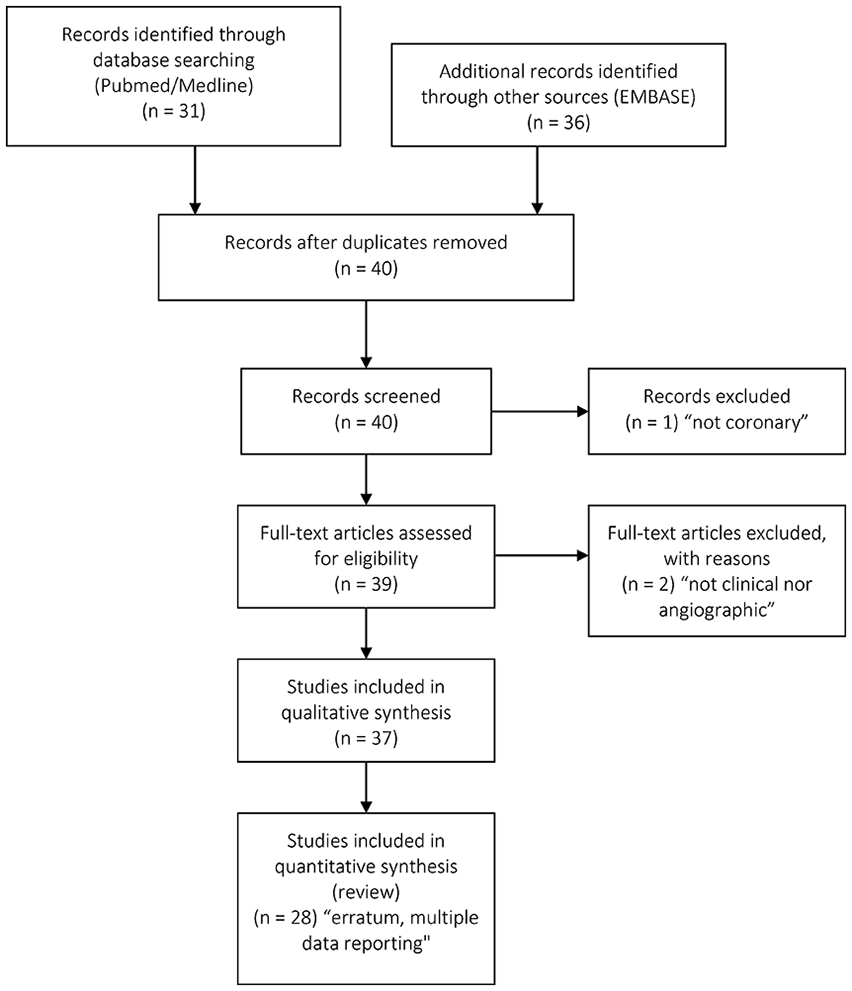
PRISMA flow chart for ‘non inferiority margin drug-eluting stent’.
NI margins of clinical endpoint trials.
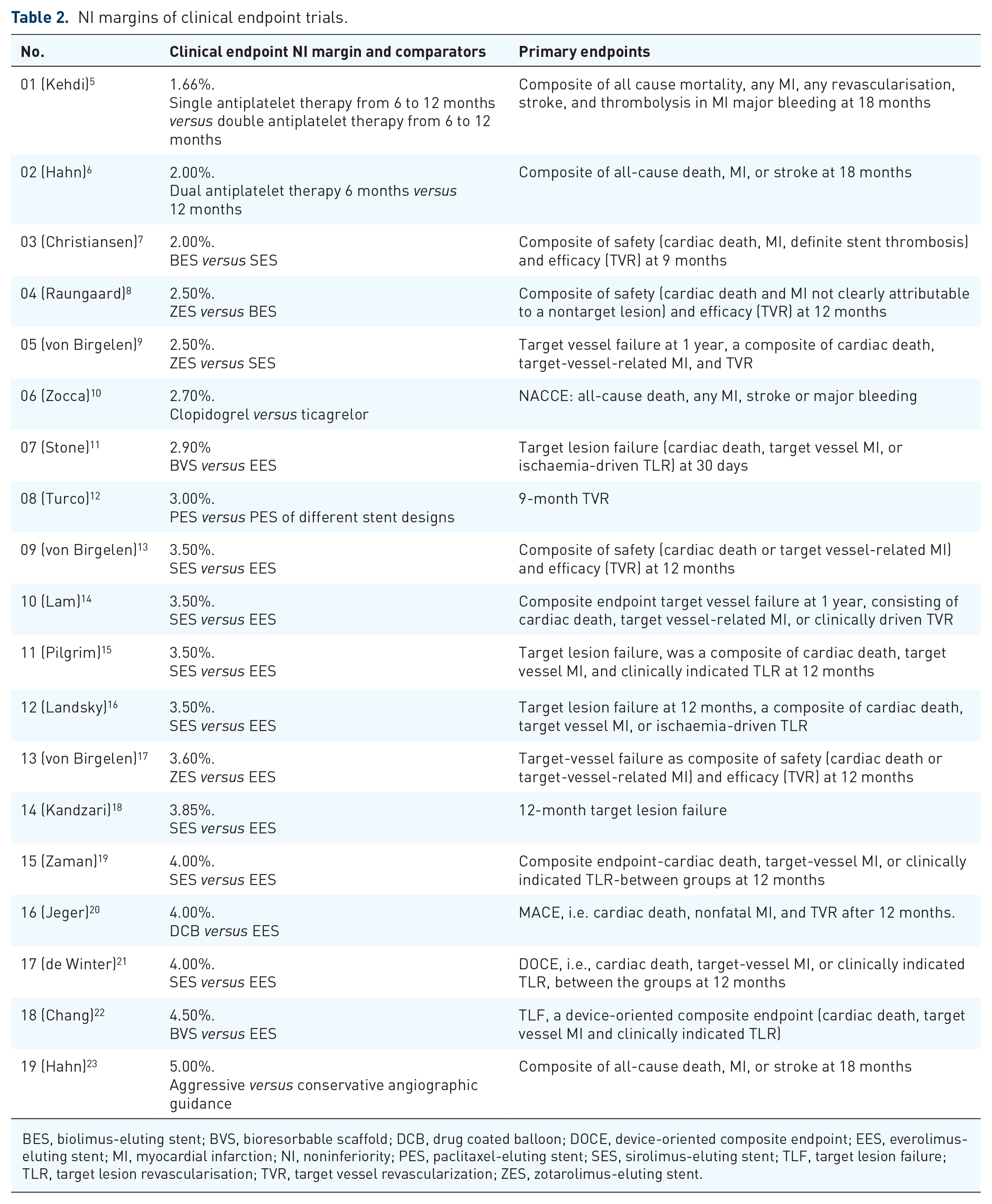
BES, biolimus-eluting stent; BVS, bioresorbable scaffold; DCB, drug coated balloon; DOCE, device-oriented composite endpoint; EES, everolimus-eluting stent; MI, myocardial infarction; NI, noninferiority; PES, paclitaxel-eluting stent; SES, sirolimus-eluting stent; TLF, target lesion failure; TLR, target lesion revascularisation; TVR, target vessel revascularization; ZES, zotarolimus-eluting stent.
NI margins of surrogate endpoint trials.

BR, binary restenosis rate; BVS, bioresorbable scaffold; DCB, drug coated balloon; DS, diameter stenosis; EES, everolimus-eluting stent; FFR, fractional flow reserve; iFR, instantaneous wave free ratio; LLL, late lumen loss; MI, myocardial infarction; PES, paclitaxel-eluting stent; SES, sirolimus-eluting stent; ZES, zotarolimus-eluting stent.
NI trials with clinical endpoints
A total of 29 recently published trials were available for analysis, with 20 references reporting clinical endpoint NI margins ranging from 1.66% to 5.00% (Table 2). The lowest NI margin of 1.66% was reported by Kedhi and colleagues. 5 They investigated different antiplatelet therapy (DAPT) strategies after event-free survival at 6 months with a composite endpoint of all cause mortality, myocardial infarction (MI), any revascularization including thrombolysis, stroke, and major bleeding. Their treatment group consisted of 432 (single antiplatelet therapy from 6 to 12 months) and 438 (DAPT from 6 to 12 months) patients.
The recently published BASKET SMALL II trial investigated DCB angioplasty and DES in small vessel de novo lesions. 20 The sample size calculations in this study were based on a NI margin of 4%, a power of 90%, and expected MACE rates of 7% in the DCB and 10% in the DES treatment groups. Lansky and coworkers compared the target vessel failure (TVF) rates in an all-comers population receiving either everolimus eluting stents (EES) or a low dose sirolimus-eluting stent (SES). 16 Their NI margin was set at 3.5%, assuming a 7% target vessel revascularization (TVR) rate in the control group. A NI rate of 4% and event rates of 8.3% in both treatment arms were used by de Winter and coworkers to study different polymer and drug coatings. 21
In a prospective RCT comparing zotarolimus-eluting stents (ZES) with SES, 9 the NI margin was set at 2.5% while assuming TVF rates at 12 months of 6.0%.
As Byrne and Kastrati pointed out, 3 NI margins that are too large have limited value for clinical ramifications, and, therefore, future treatment recommendations. We ventured into sample size calculations with a 10% clinical event rate in the control group, various event rates in the treatment group, and a range of NI margins. Figure 2 details the patient group sizes with typical MACE/TVF rates and a range of NI margins. For a typical NI RCT, a minimal treatment group size starts at around 500 patients. A range of 250–1500 patients per group can therefore be expected (Figure 2).

Number of patients per treatment group for various NI trial designs with MACE as primary endpoints for different expected MACE rates in the treatment group (top panel) and different NI margins (bottom panel).
NI trials with surrogate endpoints
Based on a previous review, 4 we would like to briefly present the most commonly used surrogate endpoints such as LLL, optical coherence tomography (OCT) and fractional flow reserve (FFR)/instantaneous wave-free ratios (iFR). Moreover, the concept of intrapatient randomization to eliminate patient-level bias appears to be of importance.
Late Lumen Loss
NI margins for LLL were reportedly used from 0.14 mm to 0.24 mm.24–29 Based on these previously published NI margins with the primary endpoint LLL, a number of common sample sizes were calculated given the expected SD, which may range from 0.40 mm to 0.70 mm. 37
Assuming a frequently reported SD of 0.45 mm, and NI margins of 0.05–0.20 mm, a minimum of 108 patients per group would have to be included given a 90% power (Figure 3). The patient population size may increase to the 1200–1700 range with a NI margin of 0.05 mm.

Number of patients per treatment group for various NI trial designs with late lumen loss as the primary endpoint.
Intrapatient randomization
There were two reports on intrapatient randomization. It was hinted that one way to increase statistical power while keeping patient numbers moderate is the use of intrapatient randomization. Intrapatient comparisons of angiographic changes in treated and untreated lesions have been previously conducted by Kleber and coworkers. 38 They studied lumen enlargement after DCB angioplasty, and compared the corresponding mean lumen diameters with those in untreated nontarget vessels. This methodology was recently applied for comparative DES studies in the FRIENDLY OCT trial (ClinicalTrials.gov Identifier: NCT02785237), in which patients with two distinct lesions received both study devices, thus patient risk factors were identical in both device groups.
The use of paired t tests to test for differences was investigated and illustrated in Figure 4. With LLL SD in the 0.40–0.45 mm range, LLL means of 0.200 mm (treatment group) and 0.250 mm to 0.325 mm in the control group, the number of patients can be reduced by half.

Number of patients per treatment group for unpaired and paired t-tests with late lumen loss (LLL) as the primary endpoint. Assumptions: LLL = 0.200 mm in the treatment group, various LLL estimates in the control group (0.250–0.400 mm) and LLL SD of 0.400 mm in both groups with a power of 80%.
New surrogate endpoints
Without an exhaustive literature search, it seems that OCT- and FFR-derived endpoints are becoming more popular and are worth mentioning. Since OCT endpoints are not the focus of this review, only a brief excursion is intended to illuminate this surrogate endpoint which is increasingly being used.39–43 Three different criteria for a ‘covered stent strut’, namely >0 µm, >10 µm and >20 µm (Table 4) have been reported in the literature.
Stent strut coverage criteria.

BP-SES, bioresorbable polymer sirolimus eluting stent; PP-EES, permanent polymer everolimus eluting stent; PF-SES, polymer-free sirolimus eluting stent.
With a range of 85–95% of covered struts at follow up, a 5% SD, sample size calculations can be made accordingly (Figure 5).

Number of patients per treatment group to test for differences in stent coverage rate using OCT with 85% in the control group and 87.5–95.0% in the treatment group with 6.0% SD in both groups.
Chevalier and colleagues, Kim and colleagues, and Suwannasom and colleagues used the criterion of >0 µm strut coverage with a 3-month follow-up.39–41 Yano and colleagues proposed a >10 µm cut-off value, 42 whereas Koppara and colleagues utilized the criterion of >20 µm. 43 Figure 5 reveals that, for strut coverage rates of 85% in the control group and, for example, 90% in the treatment group, 32 patients are necessary to detect a differences in stent coverage rates (power = 90%, SD = 6%).
FFR measurements translate into pressure drops across lesions. The higher the degree of stenosis, the higher the pressure drop. While this relationship is nonlinear, it serves as an accepted diagnostic tool to determine the severity of the lesion. Needless to say, the amount of myocardium affected by a stenosed segment is of cardinal importance to determine the need for revascularization. Shin and coworkers investigated FFR at 9 months after DES implantation or DCB angioplasty. 44 They used an unpaired t test design to demonstrate that the FFR values between treatment groups were not different (0.86 ± 0.06 versus 0.83 ± 0.08, p = 0.105). Alternative study designs such as a loss in FFR analogous to a loss in lumen diameter beyond their statistical theory were not reported in clinical study reports so far. 4 Recently, iFR have been used, 41 which do not require intracoronary administration of pharmacological vasodilators.
Table 3 lists NI margins for FFR and iFR trials. The range from 0.030 to 0.050 was used to conduct sample size calculations for this hemodynamic endpoint.33–36
Very common SD for iFR and FFR are 0.09–0.10 with clinically relevant cut-off values depending on the type of FFR.33,45 Based on the aforementioned SD, which are more on the conservative side, and NI margins, sample sizes range between 64 and 235 per group (Figure 6). Comprehensive reviews on iFR and FR were recently provided by Baumann and coworkers.46,47

Number of patients per treatment group to test for differences in iFR and FFR with a common SD of 0.05 and various NI margins of 0.020, 0.025, 0.030, 0.035, and 0.040 and power of 80% and 90%.
New clinical endpoints
Despite the fact that, to the best of the authors’ knowledge, there have been no reports with ST rates as primary endpoints, this potential endpoint is worth mentioning. There is sufficient interest for some novel DES to reduce the DAPT. If one ventures to delve into sample size calculations with the primary endpoint rate of ST with a comparator rate of 0.5% and expected rates in the treatment group between 0.4% and 0.2%, patients will be typically in the ten thousands to single digit thousands per treatment group (Figure 7). In this scenario, a composite endpoint including TVR and nonfatal MI, potentially complemented with rates of bleeding complications, may be the only feasible option to reduce the sample size to manageable numbers. Christiansen and colleagues studied the safety and efficacy of biolimus-eluting stents (BES) versus sirolimus-eluting stents (SES) by adding the rates of ST to their MACE endpoint. 7 Compared with other designs for device trials, their NI margin was only 2.0% with patient group sizes of 1208 and 1193, respectively.

Number of patients per treatment group for various trial designs to test for differences in the rate of ST with 0.5% in the control group and rates from 0.20% to 0.40% in the treatment group.
Discussion
An important aspect before feasible study designs are discussed, one ought to critically reflect whether differences in event rates or morphometric variables translate to clinical benefits for the patient. If we measure LLL in hundredths of millimeters with SD in the range or even higher than their respective mean values, what is the measurable benefit for the patient? We strongly believe that there needs to be a balance between statistical and clinical significance.
NI designs
The majority of clinical NI study designs use the classical MACE endpoint in most device trials.4,7,9,10–18 The concept of MACE is complemented by bleeding and stroke rates whenever treatment groups involve different DAPT regimens (drug regimens or durations).5–6,10 With the exception of Hahn and coworkers, 23 who investigated different procedures for revascularizations by choosing the combination of MACE and ST, there seem to be two clusters of study endpoints, namely MACE for device trials, and the combination of MACE, bleeding, and ST rates for DAPT studies. This appears very reasonable given the challenge to counterbalance the risks of ischemic versus bleeding events. Our assessments reveal that, with the addition of bleeding and ST rates to MACE rates, NI margins can be reduced to the 2.0% range5–6,10 with acceptable patient group sizes of 432 patients 5 , 1355 patients, 6 and 1009 patients. 10
An aspect of paramount importance is justification of the NI margin, which has been adequately addressed in the excellent review papers by Head and colleagues and Rehal and colleagues.48,49 While our sample size estimates (Figure 2) are based on a clinical event rate of 10% with NI margins from 1.5% to 5.0%, this may be interpreted as overly simplistic, since most clinical event rates for a composite endpoint such as MACE at 12 months may be as low as 4.5% with a NI margin of 2.5%. 9 The relevant United States Food and Drug Administration (FDA) guideline cautions the trialist to choose a NI margin greater than the actual treatment effect. 50 This will lead to a false conclusion that the new treatment is effective as compared with the control group.
Intrapatient randomization
As previously reported, intrapatient randomizations enable the clinical trial designer to eliminate all bias-introducing factors relative to patient demographics. However, in terms of lesion morphological factors, there may still be some differences. The use of the paired t test may provide some additional power, while the number of patients could be greatly reduced for proof of concept studies. 4 Overall, as can be seen in Figure 4, the patient population could be reduced to half if the study design permits two treatments per patient, for example two different stents in one patient.
Surrogate endpoints
A simplified overview of commonly used endpoints is given in Figure 8. This review did not focus on hemodynamic endpoints such as FFR and iFR. Nevertheless, we used common estimates to provide a range of study population sizes. As mentioned earlier, these are pressure gradient measurements across lesions that can either guide clinical decision making or could be even used as an endpoint measure. 35 Shin and coworkers used FFR as a surrogate endpoint at 9 months after DES implantation or DCB angioplasty. 44 They found FFR measurements after predilatation with an uncoated balloon useful as a decision facilitator. Shin and colleagues also concluded that FFR may be an option to avoid DES stenting in case long-term antiplatelet therapy is not well tolerated by the patient.

Overview of commonly used endpoints in coronary intervention trials and patient group sizes per treatment group.
OCT endpoints may be an attractive option for morphometric measurements of stent healing. From a statistical viewpoint, the >20 µm criterion appear to be advantageous since the absolute stent coverage rates are somewhat lower as compared with the other criteria. Hence, the total number of patients will be somewhat lower given a similar SD of 6%. This remains debatable if true tissue coverage and the precision of OCT are in agreement with stent coverage of less than 20 µm.
Conclusion
Acceptably low NI margins can be used with expanded composite clinical endpoints, which are MACE complemented by ST and bleeding rates. In addition to expanded clinical endpoints, intrapatient randomization may be a suitable tool to meet future needs in device approval, recertification, and reimbursement.
Study limitations
The presented sample size estimates provide the junior trialist with a first ‘stepping stone’. They do not replace calculations for a given study design. Moreover, study endpoints, that is long-term safety versus proof of concept, are not interchangeable. It was not the intention to delve into all aspects of NI trial designs, and critical aspects such as missing data, type I error rates, and sensitivity analyses were not within the scope of this review.48–50