
Abstract
Cardio-oncology is a new and rapidly expanding field that merges cancer and cardiovascular disease. Cardiovascular disease is an omnipresent side effect of cancer therapy; in fact, it is the second leading cause of death in cancer survivors after recurrent cancer. It has been well documented that many cancer chemotherapeutic agents cause cardiovascular toxicity. Nonetheless, the underlying cause of cancer therapy-induced cardiovascular toxicity is largely unknown. In this review, we discuss the potential role of damage-associated molecular patterns (DAMPs) as an underlying contributor to cancer therapy-induced cardiovascular toxicity. With an increasing number of cancer patients, as well as extended life expectancy, understanding the mechanisms underlying cancer therapy-induced cardiovascular disease is of the utmost importance to ensure that cancer is the only disease burden that cancer survivors have to endure.
Keywords
Introduction
Cancer therapy has advanced dramatically in such a way that not only do cancer patients have increased life expectancy, they also have increased survival rates. 1 Currently, there are 15.5 million patients living beyond a cancer diagnosis and by 2026, that number is estimated to increase to 20.3 million. 1 However, stemming from both short- and long-term cancer treatment, which is often given in multi-drug combinations and escalating doses, new problems are emerging. 2 One such problem is cardiovascular complications, which is the second leading cause of death in cancer survivors, only after cancer recurrence, cancer progression, and secondary malignancies. 3 In fact, cardiovascular complications have become so prominent that a new area of research is emerging within both cardiology and oncology to prevent and treat cancer therapy-induced cardiotoxicity; this field is referred to as cardio-oncology.
Interestingly, several studies have demonstrated a poor correlation between traditional cardiovascular risk factors such as obesity, dyslipidemia, insulin resistance, and tobacco use, with the onset of cardiovascular disease in cancer survivors,4,5 suggesting an alternative mechanism for systemic cardiovascular toxicity. One such mechanism could be inappropriate immune system activation, which has an active role in both cancer and cardiovascular disease.6–9 It has been well established that the immune system not only responds to foreign substances (i.e. pathogens), it also responds to endogenously derived molecules that are expressed as a result of tissue damage or stressed cells, known as damage-associated molecular patterns (DAMPs).10,11 DAMPs are present in both cancer and cardiovascular disease; however, the roles that DAMPs play are not congruent. In cardiovascular disease literature, high and chronic levels of DAMPs have been shown to be inflammogenic and are associated with disease pathogenesis.12,13 In cancer, the presence of DAMPs has been shown to be a sign of both treatment efficacy and the development of resistance.14,15
Mechanisms of action for therapies such as chemotherapy and radiation have been proposed and reported in the literature;16–21 however, due to the underlying goal of cancer therapy to reduce tumor size, we hypothesize that the unintentional release of DAMPs after any cancer therapeutic contributes to the development of cardiovascular disease, including hypertension. This review describes the increased prevalence of cancer therapy-induced cardiotoxicity after cancer therapy and the possible role that DAMPs play in the disease process.
Cancer therapies cause cell death
Cancer is a complex disease due to the ability of cancer cells to adapt to changing environmental conditions guaranteeing growth and survival. Cancer is the result of one or more DNA mutations, genetically predisposed or environmentally acquired, that allow cells to escape the normal mechanisms constraining cell division and growth. Regardless of mutation or cancer type, cancer cells exhibit four main characteristics, (1) uncontrolled proliferation, (2) de-differentiation and loss of function, (3) invasiveness, and (4) metastasis. Cancer therapies are designed to target these abnormal characteristics.
Cancer therapy type and regimen are often determined by factors specific to the patient and tumor characteristics, with the intention to cure, prolong survival, or relieve symptoms. The classic dogma of cancer therapy uses a continuous dosing strategy to ‘kill as many cancer cells as possible’. In this type of treatment strategy, there is a delicate balance amid median effective dose (ED50), lethal dose (LD50), toxicity, and drug resistance. 22 Currently, there are research groups and clinical trials studying differing dosing schedules aimed at thwarting tumor evolution. 22 Using effective treatment modality, dose, and frequency are several ways clinicians combat this multifaceted disease. 22
The main treatment categories for cancer include surgery, radiation, chemotherapy, hormone therapy, immunotherapy, and targeted therapy, which can be mixed and added together for augmented outcomes. Excluding surgery, which entails the resection of a malignant tumor, all other forms of cancer treatment aim to destroy the pro-proliferative and de-differentiated cancer cells within the body.
Radiation is a form of cancer therapy used mainly against solid tumors. Radiation utilizes high-energy particles or waves to kill cancer cells by inducing irreparable DNA breaks within cells. This method targets rapidly dividing cells; however, its effects may not take place until days or weeks after initiation and cell death could continue months after treatment is completed. Chemotherapy is another form of cancer therapy, which utilizes cytotoxic drugs to treat disseminated cancer; solid tumors are not as sensitive to this treatment option. Chemotherapy targets rapidly growing and dividing cells, and slows tumor proliferation by inhibiting specific aspects of cellular replication, ultimately leading to cell death. As the name implies, targeted therapies are more specific when compared with chemotherapy, and are directed against specific molecules and pathways that drive tumor growth. 23 For example, these types of therapies are normally small molecule inhibitors or monoclonal antibodies that block or turn off chemical signals, modify certain proteins, trigger the immune response, or carry toxic substances to cancer cells, thus resulting in cancer cell death. Targeted therapies often lead to tolerance due to overexpression of the targeted protein or through acquired mutation making the drug useless. 24 Immunotherapy reinforces or sensitizes the immune system as a defense against tumors. The main categories of immunotherapy include monoclonal antibody therapy (also considered a targeted therapy), immune checkpoint inhibitors, cancer vaccines, and other nonspecific immunotherapies. 25
While there is no doubt cancer therapies are effective at targeting and eliminating cancerous cells within the body and increasing patient survival, 1 these treatments also induce unpleasant and unintended side effects. These side effects not only include those that affect quality of life (e.g. drowsiness, restlessness, anxiety, loss of libido, etc.), they also promote other life-threatening complications, including cardiovascular disease.3,26 Nonetheless, the mechanisms underlying cancer therapy-induced cardiotoxicity remain relatively unexplored.
Clinical definitions and guidelines for cardiotoxicity after cancer treatment
The National Cancer Institute (NCI) defines cardiotoxicity as ‘toxicity that affects the heart’. 27 Aside from this broad definition, there are no universal guidelines for identifying and measuring cancer therapy-induced cardiotoxicity. 28 As a result, multiple interpretations of the NCI definition exist. For instance, the NCI Common Terminology Criteria for Adverse Events (CTCAE) 29 is considered the standard for clinical trial reporting of adverse events, 30 and it includes left ventricular dysfunction (LVD) and heart failure (HF) within its terminology, but does not include hypertension. However, other entities vary in their cardiotoxicity definition. For instance, the Food and Drug Administration (FDA) defines anthracycline cardiotoxicity as >20% decrease in left ventricular ejection fraction (LVEF) when baseline LVEF is normal, or >10% decrease when baseline LVEF is not normal.28,31 Multiple other studies, including the Herceptin Adjuvant (HERA) trial and the Breast Cancer International Research Group (BCIRG), use different measurements to define cardiotoxicity, although LVD is frequently cited.32–34
Plana et al. 35 offer a more structured classification system for cancer therapy-induced cardiotoxicity, grouping symptoms into two groups, type 1 or type 2. The type 1 category of cardiotoxicity uses doxorubicin as the representative agent and encompasses cancer therapies with dose-dependent side effects and induces irreversible myocardial damage. Type 2 uses trastuzumab as the typical agent: this classification describes cancer therapy side effects that are not dose related and are reversible. Symptoms in each category can then be further classified as symptomatic or asymptomatic. The various definitions and classifications of cardiotoxicity, paired with the lack of knowledge of cancer therapy-specific pathophysiological mechanisms have made it difficult for clinicians to detect and therefore prevent and manage cancer therapy-induced cardiotoxicity.35,36
Regardless of a lack of universal classification guidelines, there are some established prevention measures in place to combat cancer therapy-induced cardiotoxicity. For example, the American College of Cardiology and American Heart Association have together established procedures for chemotherapeutic agents and radiation therapy that have well described cardiovascular side effects, in order to reduce/prevent them.37–40 To combat these pathologies, the first line of defense is traditional cardioprotective therapies (e.g. iron chelators, angiotensin-converting enzyme inhibitors (ACE-I), beta-blockers, and statins). 41 In fact, dexrazoxane, an iron chelator, has been found to be cardioprotective in women diagnosed with advanced breast cancer and receiving anthracycline-based chemotherapy.39,42 Statin therapy has been noted to decrease heart failure incidence in breast cancer patients; 43 however, given the potential side effect of rhabdomyolysis, statins should be used with caution.43,44 While the use of these cardioprotective agents seems to reduce cardiotoxicity, 41 the need for universal guidelines for cardiotoxicity identification, as well as novel prevention and treatment options, is emphasized by the high rates of cardiovascular morbidity and mortality following cancer therapy. 2
Evidence for cardiovascular complications after cancer treatment
In the United States, an estimated one in three adults has been diagnosed with hypertension, 15 million people live with cardiovascular disease (e.g. heart failure, arrhythmias, cardiac dysfunction, etc.), 45 and approximately 14 million individuals have a prior history of cancer. 9 As the US population ages and cancer prognosis improves, so will the number of patients living with both cardiovascular disease and cancer. Not only is cardiovascular disease a leading cause of death among cancer survivors, there is an elevated risk of developing cardiovascular pathology which approaches 40% of the cancer population. 36 The elevated risk of developing cardiovascular disease could be confounded by pre-existing genetic, lifestyle, age, endocrine, and environmental factors unique to each cancer patient (Figure 1). However, these factors, as well as traditional cardiovascular disease risk factors (obesity, dyslipidemia, insulin resistance, and tobacco use),4,5 do not fully account for the increased incidence of cancer therapy-induced cardiovascular toxicity. 18

Common risk factors that contribute to both cancer and cardiovascular disease. Adapted from Irvine Page’s Mosaic Theory of Hypertension. 46
The main cancer therapies reported to induce cardiovascular dysfunction and disease are radiation, vascular endothelial growth factor (VEGF) inhibitors, which encompass tyrosine kinase inhibitors (TKIs) sorafenib and sunitinib, as well as monoclonal antibodies bevacizumab and ramucirumab, human epidermal growth factor receptor type 2 (HER2) monoclonal antibody trastuzumab, and chemotherapeutic agents such as anthracyclines, platinum-based antineoplastic drugs, microtubule inhibitors, and antimetabolites. Cardiotoxicity induced by these agents could be the result of either on-target effects (e.g. tyrosine kinases located on noncancerous vascular cells) or off-target effects (e.g. implicating pathways of vascular cells differently from cancerous cells) of cancer therapies. 23 The time-dependent effects of cancer therapy-induced cardiotoxicity vary according to the specific therapy and the patients’ baseline measurements. Table 1 lists the major forms of cancer therapy and summarizes their reported cardiovascular side effects, as well as their proposed mechanisms.
Common cardiovascular side effects of cancer therapy and potential mechanisms leading to dysfunction.

ABL1, Abelson tyrosine-protein kinase 1; ATP, adenosine triphosphate; DNA, deoxyribonucleic acid; ECG, electrocardiogram; HER2, human epidermal growth factor receptor 2; IFN, interferon; KIT, stem cell factor receptor/CD117; LVD, left ventricular dysfunction; LVEF, left ventricular ejection fraction; NO, nitric oxide; PDGFRα, platelet-derived growth factor alpha; PRR, pattern-recognition receptor; RNA, ribonucleic acid; VEGF, vascular endothelial growth factor.
Radiation
Radiation is an effective cancer therapy, nevertheless it has serious cardiovascular side-effects, such that it has been termed radiation-induced heart disease (RIHD) and radiation-induced vascular disease (RIVD).20,21 These symptoms include pericarditis, cardiomyopathy, myocardial fibrosis, coronary artery disease, peripheral vascular disease, pericardial effusion, valvular disease, arrhythmias, and autonomic dysfunction.20,47 RIHD can have acute, subacute, and late presentation and may affect the entire cardiovascular system at any stage. 21 Besides the well-known radiation effects to the heart, medium-sized to large-sized vessels demonstrate lipid accumulation, inflammation, thrombosis, increase in intimal thickness, and connective tissue after exposure.91,92
Cardiotoxicity, secondary to radiation, is increased in severity and incidence with increasing doses of radiation (i.e. dose dependent), volume of body area exposed, time since exposure, and adjuvant therapies. 19 The cancer therapy-induced cardiotoxicity effects of radiation have been observed 15–30 years after completion of therapy. 93 The location of exposure has also been reported to affect cardiotoxicity. For example, studies show that radiation over the left side of the chest, compared with the right, results in increased occurrence of cardiovascular pathology. 94 In addition, radiation exposure of the mediastinum and neck causes higher rates of cerebrovascular disease and hypertension, which could be mediated by carotid baroreceptor damage.95,96 Known mechanisms of cardiotoxicity from radiation exposure comprise fibrosis, endothelial cell damage, 20 and oxidative stress. 48
Antiangiogenesis
Angiogenesis refers to the formation of new blood vessels, which can contribute to tumor growth and survival.97,98 There are two classes of antiangiogenesis therapies for the use of cancer therapeutics: (1) antibodies specific for VEGF (e.g. bevacizumab) and (2) small molecular tyrosine kinase inhibitors (TKI) against the VEGF receptor (e.g. sorafenib and sunitinib). Although VEGF contributes to the development of cancer via the formation of new blood vessels in tumors, it also has an important role in the normal physiologic function of endothelial and renal cell survival, vasodilation, and cardiac contractile function. 99 Therefore, its inhibition has significant cardiovascular side effects. Specifically, VEGF inhibition induces conditions such as hypertension, thromboembolism, ischemia, cardiac contractile dysfunction, and heart failure. 78 A retrospective analysis of clinical trials demonstrated elevated blood pressure within 4 weeks of initiating therapy with sunitinib. 81 Mechanisms that contribute to these pathophysiological states include, inhibition of nitric oxide (NO) and prostacyclin, increased production of endothelin-1, 79 oxidative stress, and cell apoptosis. 80 It is important to note that the approval for the use of bevacizumab was revoked in 2014 for the treatment of breast cancer due to high incidence of heart failure. 75 Interestingly, VEGF tyrosine kinase inhibition has an increased rate of cardiovascular toxicity, when compared with VEGF immunotherapy due to poor selectivity of the drugs and off-target effects. 100
Tyrosine kinase inhibitors
Due to the ubiquitous nature of tyrosine kinase receptors, the class of TKI targets several different pathways. For example, sunitinib and sorafenib target VEGF, lapatinib targets epidermal growth factor receptor (EGFR) and HER2 (see below), and imatinib targets Bcr-Abl tyrosine kinase. Inhibition of each of these tyrosine kinase receptors exerts a unique cardiovascular pathology that includes heart failure,76,101,102 cardiomyopathy,73,74 hypertension, 78 thromboembolism, and cardiac contractile dysfunction. In addition to mechanisms of VEGF inhibition-induced cardiovascular disease (see above), TKIs against VEGF have also been shown to disrupt mitochondrial metabolism, impair myocardial function, and result in myocardial apoptosis. 81 These cardiovascular toxicity events have been shown to persist for years after therapy cessation. 103
Human epidermal growth factor receptor type 2 immunotherapy
The use of monoclonal antibodies such as trastuzumab requires routine monitoring for cardiac function. Trastuzumab is targeted against HER2, a tyrosine kinase receptor that mediates cell growth and survival. This type of cancer therapy has been shown to induce cardiac contractile dysfunction, reduction in left ventricular function, heart failure, hypertension, and increased sympathetic tone.49,57,85–88 Inhibition of HER2 via trastuzumab is associated with the development of cardiotoxicity as early as 4–8 weeks after initiation of therapy, albeit reversible through treatment cessation.104,105 The cardiac myocyte HER2 receptor is activated when bound with neuregulin 1-β resulting in the promotion of protein synthesis, cell survival, and hypertrophy. 106 The mechanisms of cardiotoxicity are hypothesized to occur through (1) disruption of cardiac repair and myocyte homeostasis and (2) increased sympathetic tone and activation of cardiac β-adrenergic receptors. 107
Chemotherapeutics
Despite efficacy at reducing cancer, the use of anthracyclines is restricted by cardiotoxicity. Anthracyclines are a class of antibiotics that inhibit topoisomerase activity by intercalating between DNA base pairs, leading to DNA damage and eventual apoptosis. 108 Cardiotoxicity occurs both after cumulative short- and long-term anthracycline exposure causing cardiac myocyte and endothelial cell injury.109–112 In a retrospective study of cancer patients treated with anthracyclines, the development of cardiotoxicity was positively correlated with patient age, treatment frequency, and dosage. 59 Low dose, standard-therapy breast cancer treatment with anthracyclines resulted in up to 20% of patients developing cardiac systolic dysfunction within 6 months of treatment. 113 Symptoms of cardiotoxicity associated with anthracycline use include cardiomyopathy, arrhythmia, acute myocarditis, 57 heart failure, 58 left ventricular dysfunction, 59 and cardiac ischemia.60,61 Identified mechanisms that underlie anthracycline-induced cardiotoxicity include: (1) oxidative damage to cardiac myocytes accompanied by lipid peroxidation and mitochondrial dysfunction, (2) modulation of topoisomerase activity and DNA damage, (3) alterations in multidrug-resistant efflux proteins, and (4) decreased mesenchymal and circulating progenitor cells.62,112,114–116
Alkylating agents target rapidly growing cells for cell death by adding alkyl groups to DNA, thereby cross-linking the strands, resulting in the prevention of replication. As a consequence of off-target effects, this class of therapeutics also induces significant cardiotoxicity. Patients undergoing platinum-based drug treatments have been reported to develop hypertension, myocardial ischemia, thromboembolism, cerebrovascular disease, endothelial dysfunction, and coronary artery disease.18,117 Furthermore, patients treated with agents such as cyclophosphamide and ifosfamide have been observed to develop heart failure, reduction in left ventricular function, and arrhythmias.118,119 Nonetheless, precise mechanisms of cardiovascular toxicity are less clear. Studies have reported increased levels of intracellular adhesion molecule-1 (ICAM-1) and plasminogen activator and inhibitor type 1,53,120 as well as exacerbated oxidative stress-induced cardiac cell death. 51 However, it has been shown that cisplatin is not completely excreted from the body and patients that exhibit late vascular toxicity also have measurable platinum serum levels years after completion of therapy, which could contribute to toxicity. 121
Microtubule inhibitors target and disrupt microtubule structure and function, preventing cell separation during cell division, thereby inducing cell death. Microtubules not only assist with separation during cell division, they also pro-vide cytoplasmic structure. Microtubules exist dynamically between a monomeric and polymeric form through a constant state of polymerization and depolymerization. Paclitaxel is a microtubule inhibitor, which binds to tubulin and inhibits depolymerization. Patients undergoing treatment with paclitaxel have shown increased rates of arrhythmias, 70 myocardial ischemia, 69 and coronary spasm. 49 The mechanism underlying this cardiovascular toxicity is largely unknown; however, paclitaxel has been shown to modulate calcium handling in cardiac myocytes. A study by Paul et al. (2000) demonstrated that depolymerization of microtubules increased calcium levels within the cytoplasm. There are also studies showing that disruption of the microtubule network affects cellular contraction. Chitaley and Webb (2002) demonstrated that enhanced microtubule depolymerization enhances aorta contraction in a Rho-kinase dependent manner. 122 Nonetheless, the clinical implication of increased vascular contraction and whether it contributes to cardiovascular dysfunction in cancer survivors remains to be determined.
Antimetabolites have an increased incidence of cardiotoxicity that can occur early in cancer treatment and are proportional with dose and frequency. 123 Antimetabolites interfere with mitosis by substituting metabolites necessary for DNA or RNA synthesis, thus damaging cells. Common cardiovascular side effects include cardiac ischemia,54,55 angina, disruption of cardiac electrical activity, and thrombosis. 86 The underlying mechanism of these deleterious phenotypes has been proposed to be oxidative stress and endothelial cell damage. 56
In summary, cancer therapy undoubtedly contributes to cardiovascular disease pathogenesis. There are multiple hypothesized mechanisms that have been postulated to result in cardiovascular toxicity; however, these mechanisms are broad and could be a deleterious side effect of cancer treatment, as opposed to the cause. We would like to offer an alternative hypothesis whereby cancer therapy induces cardiovascular toxicity through the action of DAMPs.
Damage-associated molecular patterns and cancer
Chronic inflammation is a distinct feature of both cardiovascular disease and cancer. 9 However, the precise mechanisms that mediate this inflammation are only beginning to emerge. While infection is unequivocally linked to the pathogenesis of both cardiovascular disease 124 and cancer, 125 many patients with these diseases present sterile inflammation, or immune system activation in the absence of a pathogen. Although sterile inflammation has been proposed to arise through different mechanisms, 126 DAMPs and the danger theory of immunity have led to a paradigm shift in the understanding of not only pathophysiology, but physiology also.10,11
DAMPs are endogenous molecules that have specific functional purposes inside cells and as such, are normally compartmentalized within membranes for performance of their various tasks. However, when these endogenous molecules are exposed to the extracellular environment, either by passive diffusion after membrane rupture or active secretion during stress, they unintentionally acquire additional immunogenic properties. These functions broadly include: the presentation of danger signals to other cells in a paracrine and endocrine manner, the activation of pattern-recognition receptors (PRRs) of the innate immune system, communication to the adaptive immune system and promotion of immunological memory, and finally the facilitation of tissue repair. 127 Obviously, these functions are not all mutually exclusive and the ability of DAMPs to both promote and resolve inflammation is an evolutionarily conserved mechanism to restore immunological homeostasis. However, problems arise when the expression of DAMPs becomes excessive, chronic, and uncontrolled. We propose that disease progression ensues when the expression of DAMPs exceeds a ‘pathogenicity threshold’ (Figure 2).
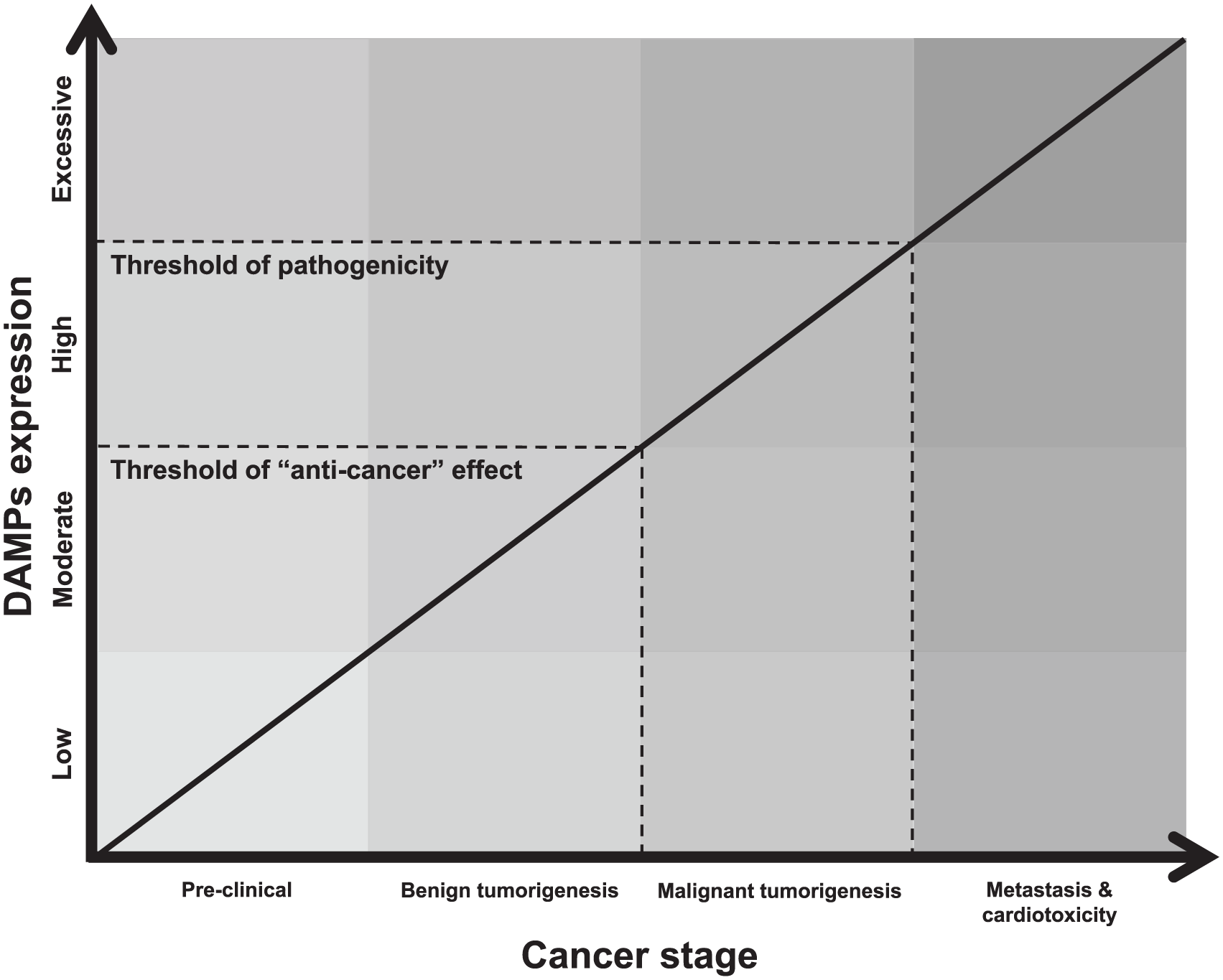
Hypothesized schematic of how damage-associated molecular patterns (DAMPs) contribute to the treatment and progression of cancer and cardiotoxicity. Due to the paradoxical contribution of DAMPs to cancer progression in the literature, we believe two key thresholds of DAMP expression exist. The first is the ‘anti-cancer’ threshold and this is where controlled amounts of DAMPs stimulate the immune system to mount an effective defense against the growing and potentially pathogenic tumor. This explains the benefits of immunogenic cell death (ICD) to cancer remission. The second threshold is the one of pathogenicity and this is when DAMP expression becomes so excessive and chronic that uncontrolled inflammation ensues, and this contributes to the progression of cancer, resistance to anti-cancer treatments, and cardiovascular disease.
There are a large number of endogenous molecules that could potentially become DAMPs, which is dependent on the extracellular environment. Currently, DAMPs include a number of macromolecules composed of lipids, nucleic acids, and proteins from different sources such as extracellular matrix and cellular organelles. To add further complexity, macromolecules released into the extracellular environment surrounding stressed or decaying cells could be modified in such a way that their pathogenicity is either amplified or abrogated (e.g. oxidation). 128 PRRs of the innate immune system (e.g. Toll-like receptors [TLRs]) are able to recognize distinct motifs within these macromolecules. Once these macromolecules are recognized, the innate immune system responds in a manner that is motif-specific and promotes a pro-inflammatory milieu that is needed to combat the danger (e.g. upon the recognition of viral pathogen-associated molecular patterns (PAMPs), the major defensive strategy employed by the host immune system is the activation of the interferon regulatory factors). Table 2 provides several examples of DAMPs observed to be released after cancer therapy, as well as their corresponding PRR. It should be noted that this list is by no means complete, as many other unknown molecules may fulfill the inclusion criteria of being DAMPs and PRR ligands, especially in the ever-changing tumorigenic microenvironment (e.g. neoantigens).
Secreted or released damage-associated molecular patterns that have been measured after various anti-cancer therapies.

ATP, adenosine triphosphate; BiP, binding immunoglobulin protein; CD, cluster of differentiation; DAMP, damage-associated molecular pattern; FEEL-1/CLEVER-1, Fasciclin EGF-like/common lymphatic endothelial and vascular endothelial receptor-1; GP96, glycoprotein 96; GRP, glucose-regulated protein; HMGB1, high mobility group box 1; HSP, heat shock protein; IFN, interferon; IL, interleukin; IL-1R, interleukin-1 receptor; LOX-1, lectin-type oxidized LDL receptor 1; PRR, pattern-recognition receptor; P2X7, purinergic receptor P2X, ligand-gated ion channel 7; P2Y2, purinergic receptor P2Y, G-protein coupled 2; RAGE, receptor for advanced glycation end products; SREC-1, scavenger receptor class F member 1; TIM3, T cell/transmembrane, immunoglobulin, and mucin; TLR, Toll-like receptor; UVC, ultraviolet C.
The participation of DAMPs as the mediators of the inflammation in most cases presumes that cell death is the initiating mechanism of their release. 151 However, determining whether cell death primarily drives pathophysiology or is a secondary bystander is difficult. Although there are many forms of cell death, 152 necrosis was originally thought of as the primary source of pro-inflammatory DAMPs due to disintegration of the plasma membrane and passive release of intracellular constituents into the extracellular environment. 153 However, we have come to learn that apoptosis can also be immunostimulatory, as a result of the programed release of immunogenic molecules,127,154 and necroptosis further refines the ability of cells to rapidly present DAMPs to the immune system with the regulated and controlled breakdown of the plasma membrane. 155 Cell stress or injury can also fuel the secretion of DAMPs 127 and this emphasizes that cell death is not an absolute precursor to participation of DAMPs in pathophysiology.
The participation of DAMPs in cancer is seemingly contradictory. On one hand, DAMPs have been proposed to play a beneficial role in cancer therapy by interacting with the immune system and promoting an ‘anti-cancer vaccine effect’ (Figure 2), even in the absence of a traditional adjuvant.156–159 On the other hand, DAMPs could contribute to the progression of cancer160–167 and promote resistance to cancer treatments.15,147 The former is made possible due to the ability of certain cancer therapies to induce immunogenic cell death (ICD) that is associated with the emission of DAMPs from dying cancer cells. DAMPs can then be trafficked by signaling pathways, which are instigated and regulated by a complex interplay between endoplasmic reticulum stress, reactive oxygen species, and certain metabolic/biosynthetic processes. 158 The ultimate response of DAMPs in cancer (i.e. antitumorigenic or protumorigenic) may depend on a number of different factors, 156 such as:
The histopathology of the cancer (and therefore its anatomical location).
The type of cell death modality that is triggered by ICD, and the biochemical processes activated (this, in itself, may also depend on a number of different and varying factors).
The types and abundance of immune cells present to phagocytose debris.
Finally, whether a cancer antigen is recognized or not.
In summation, the conflicting contributions of DAMPs in cancer have revealed a Janus face.157,159 Nonetheless, given the well-documented role of DAMPs in exacerbating inflammation in cardiovascular disease, 168 we believe that DAMPs, irrespective of their contribution to cancer treatment and progression, could serve as a novel facilitator of cardiotoxicity during and following cancer therapy. 49 DAMPs in cancer could arise from a number of different sources, including (paradoxically) tumor growth and remodeling,160–167 as well as carcinogenic environmental toxins. 169
Damage-associated molecular patterns and cardiovascular disease
While it has been reviewed in more depth elsewhere, 168 there is ample evidence that increased levels of DAMPs are associated with cardio-vascular disease in human populations. For example, hypertensive patients have higher plasma levels of mitochondrial DNA, high mobility group box 1 (HMGB1), heat shock protein (HSP) 60 and HSP70, or fibrinogen, and each of these is capable of activating distinct PRRs that then start the immune-response cascade. 168 Additionally, DAMPs such as HMGB1, advanced glycation end products (AGEs), hyaluronan, oxidized low-density lipoprotein, and uric acid are present in higher levels in the circulation or the plaques in patients with atherosclerosis and S100 proteins are increased in stroke.170,171 Consequently, PRRs such as TLRs seem to be chronically activated in cardiovascular disease, including atherosclerosis, cardiomyopathy, hypertension and cerebrovascular disease.12,168,172–175
There are also strong indications from basic science studies that release of DAMPs is causative for cardiovascular disease. Thus, experimental administration of DAMPs has detrimental effects on cardiovascular parameters and inhibition of DAMPs, or their respective PRRs, improves cardiovascular disease phenotypes and outcomes. 168 HMGB1 administration increased myocardial infarct size and promoted the development of microvascular thrombosis. 176 Our group demonstrated that mitochondrial DNA infusion leads to hypertension in a pre-eclampsia rodent model. 177
Additionally, treatment with TLR4 or TLR9 inhibitors decreased blood pressure in spontaneous hypertensive rat (SHR), and treatment with the TLR4 inhibitor eritoran decreased myocardial ischemia-reperfusion injury; 12 TLR4 and TLR2 deficient mice are protected from doxorubicin-induced cardiomyopathy.178,179 Genetic deficiency in various TLRs or their common downstream signaling partner MyD88 protects apolipoprotein E (apoE) knock-out mice from atherosclerosis. 180
Hypothesis
As discussed above, there is a clear link between cancer therapy and cardiovascular disease. While it is logical to surmise that the traditional cardiovascular disease risk factors (e.g. obesity, dyslipidemia, insulin resistance, and tobacco use) contribute to cancer therapy-induced cardiotoxicity (especially given that these are also risk factors to cancer itself), correlational analysis does not support this notion.4,5 Therefore, the clinical and basic science data summarized in this review strongly support the notion that cancer therapy directly causes cardiovascular toxicity that would not have occurred in its absence. The mechanisms by which specific cancer therapies or radiotherapy lead to cardiovascular disease is in some cases clearly demonstrated, for instance interference with endothelial NO signaling in the case of VEGF receptor inhibitors. In the majority of remaining cases, however, the processes by which cancer therapy causes cardiovascular disease are unclear. We believe that DAMPs and innate immune-system activation are the missing contributing factors for the development of cancer therapy-induced cardiotoxicity (Figure 3).

Danger signaling in cardio-oncology. The nature of cancer therapy is to reduce tumor size by causing sudden and rapid cancer-cell death. Because damage-associated molecular patterns (DAMPs) are released from dead and dying cells (via necrosis or apoptosis), this results in the release of DAMPs from cells during cancer therapy. DAMPs activate the immune system through activation of pattern-recognition receptors from cell types within the cardiovascular system (e.g. endothelial cells, vascular smooth muscle); we hypothesize that the overactivation of this system results in a pro-inflammatory cardiovascular disease following cancer therapy.
In support of this hypothesis, chemotherapeutic agents and radiotherapy are effective against cancer because they induce cell death in rapidly dividing cell populations. As a consequence of sudden cell death in a large number of cells simultaneously, there is a rapid and massive release of various DAMPs that enter the systemic circulation and induce an inflammatory response through activation of the innate immune system. As discussed above, both in vivo and in vitro treatments with chemotherapeutic agents or radiation induce release of DAMPs (Table 2). An increase in circulating DAMPs post-chemotherapy could contribute to the increased prevalence of thrombosis in cancer patients. 181 Thrombosis has a physiological role in immune defense, where monocytes respond to PAMPs and DAMPs by releasing tissue factor and initiating coagulation pathways. 182 Therefore, an increased presence of circulating DAMPs could play a role in the increased presence of thrombotic events in these patients. It has also been shown that chemo- and radio-therapy causes activation of innate immune receptors, such as PRRs. 146
In a perfect illustration of the Janus face effect, DAMP release and PRR activation following administration of chemotherapy may be simultaneously beneficial for cancer treatment and detrimental for other systems and organs, including the cardiovascular system. Thus, ICD can drive both immune-mediated tumor suppression and pro-inflammatory cytokine-mediated tissue injury via DAMPs. Similarly, immune cells, and in particular, T cells, also present a paradox in the context of the pathophysiology of cardio-oncology. While promoting T-cell expansion may be beneficial in diminishing cancer progression and tumorigenesis, they are also known to promote cardiovascular disease, 183 and specifically, hypertension and its associated hallmarks. 184
The therapeutic potential of adding agonists for TLR9 (CpG DNA), TLR3 (poly I:C), or TLR4 (endotoxin analogs) for a synergistic effect to radiotherapy or chemotherapeutics was recently evaluated and clear anti-tumor effects have been ascribed to this approach. 185 On the other hand, long-term consequences of TLR ligand administration on any cardiovascular parameters are only beginning to be investigated. 186 Data from our group and others would suggest that high amounts of DAMPs, whether released from tumor cell death following cancer therapy or administered therapeutically, might have a negative impact on the cardiovascular system.
Potential value of damage-associated molecular patterns in cardio-oncology
There are continuing advances in cancer therapeutics; however, the challenge now exists of developing avenues allowing these treatments to be efficacious without significant cost. The prevalence of cancer therapy-induced cardiotoxicity has led to systematic monitoring and the need for early identification of biomarkers for high-risk patients.187,188 Currently, circulating cardiac troponins (TnI, TnT) are considered the gold-standard marker for cardiac injury and are used as a diagnostic adjuvant to echocardiograms and other diagnostic modalities. TnI is sensitive and specific for myocardial injury allowing for early myocardial damage detection prior to any clinical detection through physical examination or imaging, especially in anthracycline-based chemotherapy regimens. 189 Other studies have also looked at the usefulness of TnI and TnT as surrogate markers for myocardial damage with the use of anti-VEGF TKI chemotherapeutics.18,190,191 Nonetheless, cardiac troponins may not be the only molecules released during cardiovascular damage after an insult such as chemotherapy or radiation. Theoretically, a wide array of circulating DAMPs could be measured in cancer patients prior to, during, and after treatment. Therefore, DAMPs present an opportunity for identifying and treating cancer therapy-induced cardiotoxicity. For example, obvious therapeutic targets for DAMPs include antagonism of the PRR activated by a specific DAMP, or direct ligand neutralization, thereby reducing the inflammation that promotes cardiovascular disease. Given the beneficial effects of DAMPs in some cancers, perhaps specific PRR antagonism in cardiovascular tissues is warranted.156–159
In conclusion, the use of DAMPs as a diagnostic adjuvant (i.e. biomarker) or therapeutic drug could be a novel approach to decrease cardiovascular morbidity and prevent premature mortality from cardiovascular toxicity for the millions of patients that have successfully outlived their initial cancer diagnosis. After all, what is the point of tolerating the toxicity of cancer-therapy to survive cancer if you subsequently succumb to cardiovascular disease?
Footnotes
Acknowledgements
Special thanks to Theodora Szasz for her contributions to the outline and hypothesis section of the paper.
Funding
This work was supported by the National Institutes of Health (grant number HL-134604, Program Project Grant 2017-22).
Conflict of interest statement
The authors declare that there is no conflict of interest.