
Abstract
Background:
Therapy with HMG-CoA reductase inhibitors (statins) has been associated with a significant reduction in the number of major cardiovascular (CV) events in diabetic patients. The mechanisms by which these drugs improve cardiac status remain unclear. We assessed the effects of atorvastatin (10 mg/kg/day) on CV function in streptozotocin (STZ)-induced diabetic rats.
Methods:
Age-matched, nondiabetic rats were used as controls. Echocardiographic parameters, systolic blood pressure (SBP), endothelial-dependent relaxation, cardiac and vascular oxidative stress, perivascular fibrosis, and cholesterol levels were evaluated after a 4-week atorvastatin treatment period.
Results:
In diabetic rats, SBP was higher than in controls. Atorvastatin decreased SBP in diabetic rats by 14% (n = 10, p < 0.05), and significantly increased stroke volume, ejection fraction, and cardiac output index. Whereas atorvastatin reduced left ventricular end systolic volume (LVESV) by 50% (p < 0.05), it failed to reduce left ventricular end diastolic volume (LVEDV). Total cholesterol was higher in diabetic rats than in controls and atorvastatin was ineffective in reducing cholesterol levels. The statin, however, decreased perivascular fibrosis and media thickness, and the markers of oxidative stress malondialdehyde (MDA) and 4-hidroxyalkenals (4-HAE) in aortic homogenates from diabetic rats. In addition, atorvastatin improved endothelial function by increasing the EMAX value of the acetylcholine-induced relaxation from 53.7 ± 4.1% in untreated diabetic to 82.1 ± 7.0% in treated diabetic rats (n = 10, p < 0.05). L-NAME fully abolished this improvement, suggesting that the increased vascular relaxation with atorvastatin is NO-dependent.
Conclusions:
Whereas atorvastatin does not reverse ventricular dilatation, it does have a positive hemodynamic effect on the CV system of diabetic rats. This hemodynamic benefit is independent of cholesterol levels, and is observed concomitantly with reduced oxidative stress, vascular remodeling, and improved endothelial function. Together, these results suggest that atorvastatin decreases the workload on the heart and improves systolic performance in type 1 diabetic rats by reducing oxidative stress, vascular tone, and systemic vascular resistance.
Keywords
Introduction
Diabetes is a metabolic disorder associated with increased morbidity and mortality, and cardiovascular (CV) disease is the most common cause of death among diabetic patients [Morrish et al. 2001]. One of the major CV complications observed in these patients is diabetic cardiomyopathy. In this clinical condition, ventricular dysfunction develops in the absence of hypertension, coronary artery disease, or hyperlipidemia [Joffe et al. 1999]. The etiology of cardiac deterioration in diabetes remains unclear, although endothelial dysfunction, increased oxidative stress, and alterations in endothelial nitric oxide synthase (eNOS) and inducible nitric oxide synthase (iNOS) status have been reported in animal models and patients with hyperglycemia [Cosentino et al. 1997; Crespo et al. 2008; Santos et al. 2012].
Statins, which are inhibitors of the 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, were originally developed to reduce low-density lipoprotein (LDL) cholesterol levels. Daily administration of atorvastatin reduces the risk of the first CV event in type 2 diabetics that do not have high LDL cholesterol [Colhun et al. 2004]. Moreover, the Standards of Medical Care in Diabetes 2013 published by the American Diabetes Association recommends that statins be added to lifestyle therapy for all diabetics with overt CV disease and those without CV disease but with one or more risk factors, regardless of baseline lipid levels [American Diabetes Association, 2013]. That statins improve the CV status of diabetics, irrespective of their cholesterol levels, suggests that these drugs have pleiotropic benefits that may extend beyond their effects as antilipidemic drugs [Yamagishi and Imaizumi, 2005; Yamagishi et al. 2006]. The mechanisms underlying these beneficial effects are unknown, although it has been reported that statins improve endothelial function by promoting enhanced nitric oxide (NO) levels through increased systemic NO bioavailability [Schäfer et al. 2005] and eNOS expression [Huang et al. 2012], and reduced oxidative stress generation [Wassmann et al. 2001; Van Leuven and Kastelein, 2005]. Statins also inhibit exaggerated platelet activation in rats with congestive heart failure [Schäfer et al. 2005] and decreased intramyocardial inflammation and myocardial fibrosis [Van Lintout et al. 2007].
Atorvastatin, one of the most prescribed statins, reduces proinflamatory and prothrombotic markers in type 1 diabetics without coronary heart disease [Konduracka et al. 2008]. This statin also improves leg artery stiffness in type 2 diabetics [Shinohara et al. 2005], and appears to reduce the risk of CV and diabetes-related events in elderly Japanese patients with type 2 diabetes [Shinozaki et al. 2012]. Atorvastatin does not improve endothelial function, however, in type 2 diabetic patients with average cholesterol levels and no prior CV disease [Tantikosoom et al. 2005], and it does not prevent new-onset diastolic dysfunction in a rat model of insulin resistance [Chen et al. 2007]. Thus, even though there is evidence that statins improve endothelial and CV function, the extent of this improvement, particularly under hyperglycemic conditions, remains unestablished and controversial.
The present study assesses the effects of a 4-week atorvastatin low-dose treatment on cardiac performance in type 1 streptozotocin (STZ)-induced diabetic rats, as evaluated by echocardiography, and correlates cardiac status with systolic blood pressure (SBP), endothelial function, oxidative stress, total cholesterol levels, and perivascular fibrosis. We found that although atorvastatin does not lower cholesterol levels or prevent the development of dilated cardiomyopathy, it decreases SBP, vascular oxidative stress and perivascular fibrosis, and improves endothelial function, cardiac output, and systolic performance.
Materials and methods
Experimental animal model
A total of 40 male Sprague–Dawley rats (Hilltop, Scottdale, PA), approximately 4 weeks of age, were divided into a diabetic group and a nondiabetic group, with each group containing 20 animals. To induce diabetes, rats were fasted overnight and then injected intraperitoneally (IP) with STZ (65 mg/kg) dissolved in a 0.1 M citrate buffer (pH 4.5). Nondiabetic animals were only injected (IP) with the citrate buffer solution. Hyperglycemia was verified 24 hours after the STZ injection with an Accu-Check Active blood glucose monitoring system (Roche, Indianapolis, IN). Blood glucose levels were monitored once a week in all animals. The experiments began 4 weeks after diabetes induction. The diabetic rats never received insulin supplementation. All animals were housed in a temperature-controlled room on a 12-hour light/dark cycle. Water and food (Harlan Rodent Diet, 18% protein) were provided ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee, and adhered to the Guide and Care for the Use of Laboratory Animals published by the National Institutes of Health (USA).
Drug administration
Diabetic and control rats were subdivided into four groups (two diabetic groups, two control groups). The same day that induction of diabetes was confirmed, one diabetic group and one control group began receiving atorvastatin (10 mg/kg/day) for a 4-week period. The drug, dissolved in corn oil, was administered daily by gavage. The other two groups received only corn oil, as a placebo, by gavage. Dosage selection was based on comparable studies of diabetic rats [Ali et al. 2011]. A dose of 10 mg/kg/day may appear higher than that commonly prescribed to patients. Owing to species differences in metabolism, however, a dose of 50 mg/kg/day is needed to produce plasma concentrations in rats similar to those achieved with common atorvastatin doses in humans [Wassmann et al. 2001].
Evaluation of CV parameters using echocardiography
Treated and untreated diabetic and control rats underwent serial transthoracic echocardiographic evaluation to assess cardiac function. The echocardiographic method, which was described in detail previously [Ikeda et al. 2000], employs a Sonosite 180P, a Digital Portable Ultrasound System (Sonosite Inc., WA) equipped with a Pulsewave Doppler, and a transducer with a wide bandwidth of 7.5–9.0 MHz. The use of echocardiography in diabetic rats has been validated previously in our laboratory with a thermodilution technique, using a Cardiomax-II-R System (Columbus Instruments OH) [Crespo and Dunbar, 2003]. Rats were anesthetized by injecting sodium pentobarbital (30 mg/kg body weight [BW], IP), and oriented in a left lateral decubitus position. The transducer was gently placed over the left hemithorax to avoid bradycardia. Using the two-dimensional parasternal short-axis imaging plane as a guide, a left ventricular M-mode tracing was obtained to measure left ventricular end systolic (LVES) and end diastolic (LVED) dimensions. Heart rate (HR) was determined from the RR intervals on the LV-mode tracings. A Sitelink Image Manager (Sonosite Inc., WA) was used to record and analyze the images that provided data on left ventricular end systolic (LVESV) and diastolic (LVEDV) volumes, ejection fraction, stroke volume, and cardiac output (CO). The cardiac output index (COI) was obtained by normalizing CO to BW.
Noninvasive determination of SBP
SBP was determined by placing a pressure cuff on the tail. The cuff was inflated to a pressure of 250 mmHg, and SBP was then recorded with a piezoelectric sensor near the pressure cuff. The sensor was connected to a microcomputer system (RTBP-2000, Kent Scientific, Litchfield, CT) that processed the signals. The data were recorded and analyzed using the program LabView (National Instruments Co. Austin, TX). The mean value of five measurements, separated by 3-minute intervals, was reported for each animal.
Determination of total cholesterol levels
To determine total cholesterol levels, blood samples were obtained from rats in each experimental group. Blood was centrifuged at 5000 rpm for 5 minutes at 4°C, and the plasma was collected and stored at −70°C. Cholesterol levels were measured with a commercial kit (Cholesterol Quantitation Kit, Catalog Number MAK043, Sigma-Aldrich, St. Louis, MO), following manufacturer instructions. Briefly, 5 µl of plasma and 45 µl of Cholesterol Assay Buffer were added directly to each microplate well, followed by 50 µl of Reaction Mix. Samples were then incubated for 60 minutes at 37°C in the absence of light. After the incubation period, the absorbance was measured at 570 nm using a SpectraMax M3 Microplate Reader (Molecular Devices, Silicon Valley, CA). Cholesterol levels were quantified by constructing a calibration curve that was based on variable amounts of cholesterol that ranged from 0 to 5 µg per well.
Measurement of endothelium-dependent relaxation
To assess endothelium-dependent relaxation, animals were weighed and then anesthetized with an IP injection of sodium pentobarbital (50 mg/kg) on the same day of the experiment. When complete anesthesia was achieved, the thorax was opened, and the descending thoracic aorta was removed. Isolated aortas were placed in Krebs’ bicarbonate solution (composition: 118 mM NaCl, 2.5 mM CaCl2, 5 mM KCl, 1.1 mM MgSO4, 25 mM NaHCO3, 1.2 mM KH2 PO4 and 10 mM glucose, pH 7.4). The connective tissue adjacent to the aortic adventitia was carefully removed, avoiding damage to the smooth muscle and the endothelium. An aortic ring, approximately 5 mm in length, was obtained from the proximal segment of each aorta and used for the vascular studies.
Aortic rings were suspended horizontally between two stainless-steel wires. Whereas one wire had a static position, the other wire was connected to a force-displacement transducer (Grass, model FT03C) that was attached to a DC preamplifier (Grass, model 7P1F). The signal was analyzed with a data acquisition card (National Instruments, PC-LPM-16/PnP), and changes in isometric tension were recorded with LabView software (National Instruments, Austin, TX). The entire preparation was mounted in a two-hook, 50 ml organ chamber (Radnoti Co, Monrovia, CA) and bathed in Krebs’ bicarbonate solution (aerated with a mixture of 95% O2 and 5% CO2 at 37°C). The rings were subjected to a resting tension of 2.0 g. We determined previously that this tension is optimal for these experiments [Crespo et al. 2003; Crespo and Dunbar, 2003]. Once the optimal tension was reached, the rings were subjected to a 1-hour equilibration period.
Concentration-response curves for acetylcholine were performed to determine the effects of atorvastatin on endothelium-dependent relaxation. Aortic rings were removed from diabetic rats after 4 weeks of treatment with atorvastatin (10 mg/kg/day) and precontracted with 0.1 µM norepinephrine. When the maximal contractile plateau was reached, cumulative concentration-response curves were generated for acetylcholine (0.1 nM to 100.0 µM). Similar experiments were performed in the presence of the NO synthase inhibitor L-NAME (1m M). For each individual experiment, the relaxation induced by acetylcholine was expressed as a percentage of the relaxation relative to the maximal contraction induced by 0.1 µM norepinephrine. The maximal relaxation achieved (EMAX) by the aortic rings, and the concentration that induces 50% of maximal relaxation (EC50) for acetylcholine, were determined through a mathematical analysis of the concentration-response curves.
Histological evaluations
Histological assays were performed on similar sections of the thoracic aorta from treated and untreated rats in order to evaluate the effects of atorvastatin on perivascular fibrosis and media thickness, both of which are reliable predictors of vascular remodeling. Comparable aortic segments were sectioned at a thickness of 5 µm with a microtome and stained with hematoxylin and eosin (H&E) to evaluate media thickness, and with Azan–Mallory stain to evaluate perivascular fibrosis. Images of the sections were recorded with a high-powered digital microscope (Fischer Scientific, Pittsburg, Penn) connected to a digital camera (Motic program; DC Imaging, West Chester, Ohio) and then viewed on a computer monitor. Media thickness and perivascular fibrosis were measured at five different locations using Sigma Scan Pro 5.0 measurement software (Systat Software, Inc., San Jose, CA). The mean value of five measurements was calculated for each animal. Results (in micrometers) were normalized to BW.
Measurement of lipid peroxidation
Lipid peroxidation, an indicator of oxidative stress, was evaluated by measuring cardiac and vascular malondialdehyde (MDA) and 4-hydroxyalkenals (4-HAE) levels using a commercial kit (Bioxytech LPO-586; Oxis Research, Portland, OR), and following manufacturer instructions. Briefly, homogenized tissues from the heart and aorta were centrifuged at 4°C for 10 minutes, and the supernatant was collected and stored at −80°C. N-methyl-2-phenylindole in acetonitrile and methanesulfonic acid were added to each sample and incubated at 45°C for 60 minutes. Samples were then centrifuged and the supernatant was transferred to a cuvette. The absorbance of each sample was measured at 586 nm using a SpectraMax M3 Microplate Reader (Molecular Devices, Silicon Valley, CA). The MDA and 4-HAE concentrations of each sample were determined according to the standard curve generated in the same setting and were normalized by protein concentration, as determined by Bradford Assay.
Statistical analysis
Results are presented as the mean ± standard error of the mean (SEM) using statistical software (GraphPad-Prism 5.0, GraphPad Software, Inc., San Diego, CA). Student’s t-test was used to compare two groups and the analysis of variance (ANOVA) to compare more than two groups. Student–Newman–Keuls test for post hoc analysis was used to further evaluate significant ANOVAs. Values were considered statistically significant at p < 0.05.
Results
Table 1 compares the general characteristics of untreated and atorvastatin-treated diabetic rats with those of control rats. During the study period, BW increased less in diabetic rats than in controls (p < 0.05). Blood glucose levels in diabetic rats were higher than 400 mg/dl, ranging from 445.41 ± 24.11 mg/dl 24 hours after STZ administration to 517.76 ± 18.11 mg/dl after 4 weeks (Table 2). Atorvastatin-treated diabetic rats exhibited glucose levels and a weight gain pattern similar to that of untreated diabetic rats. Thus, the HMG-CoA reductase inhibitor did not modify these variables in diabetic rats.
Body weight (g) of control and diabetic rats treated with atorvastatin (ATV).
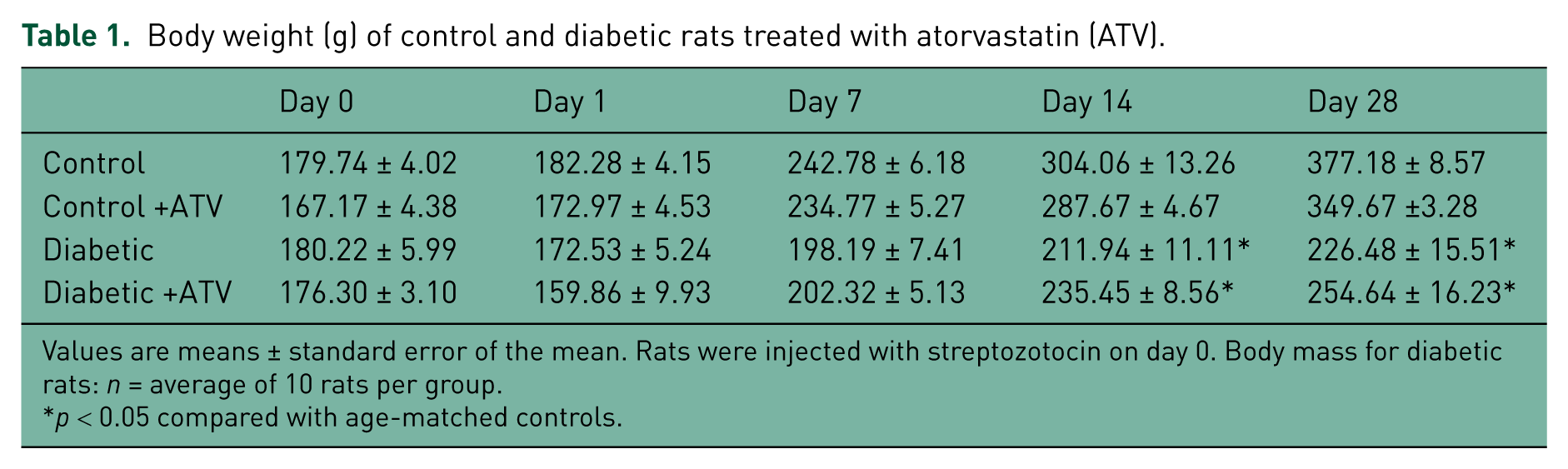
Values are means ± standard error of the mean. Rats were injected with streptozotocin on day 0. Body mass for diabetic rats: n = average of 10 rats per group.
p < 0.05 compared with age-matched controls.
Blood glucose (mg/dl) of control and diabetic rats treated with atorvastatin (ATV).
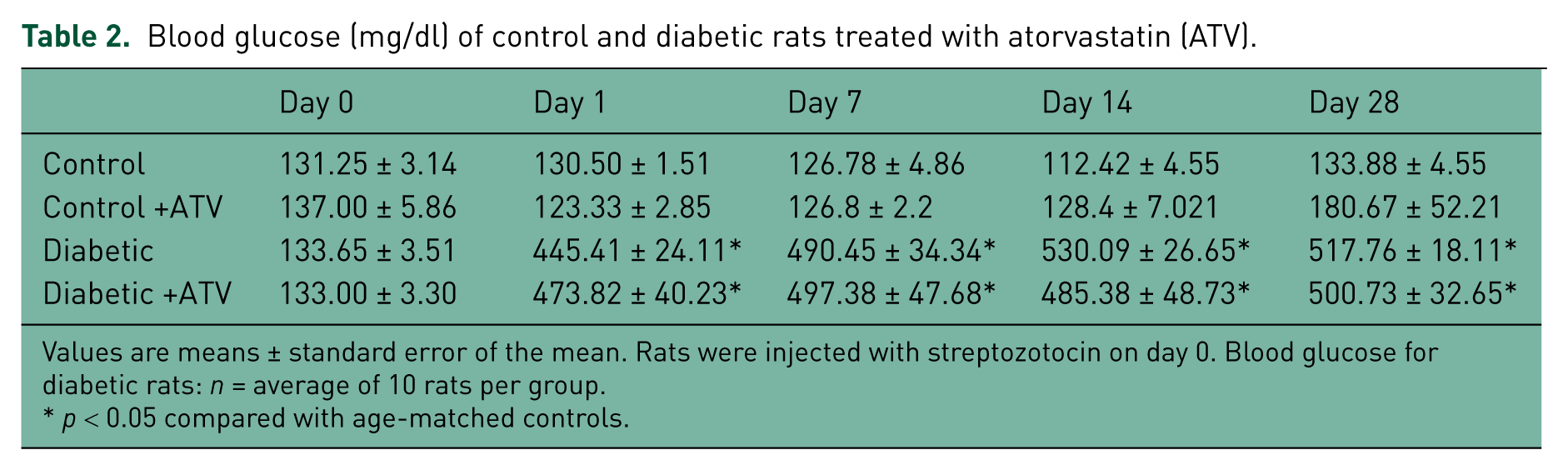
Values are means ± standard error of the mean. Rats were injected with streptozotocin on day 0. Blood glucose for diabetic rats: n = average of 10 rats per group.
p < 0.05 compared with age-matched controls.
Echocardiographic results are shown in Table 3 and in Figures 1(A) and (B). In untreated diabetic rats, heart rate, stroke volume, COI, and ejection fraction were significantly lower than in aged-matched controls. When diabetic rats were treated with atorvastatin, however, stroke volume increased from 0.20 ± 0.02 ml to 0.51 ± 0.06 ml (n = 8, p < 0.05), COI increased from 23.7 ± 3.65 ml/min/100 g BW to 57.7 ± 7 ml/min/100 g BW (n = 8, p < 0.05), and ejection fraction increased approximately 33% from 44.9 ± 3% to 59.9 ± 3% (n = 8, p < 0.05). Atorvastatin also reduced LVESV from 0.18 ± 0.04 ml/100 g BW to 0.09 ± 0.02 ml/100 g BW (n = 8, p < 0.05; Figure 1(A)). In addition, dilated cardiomyopathy was evident in diabetic rats, as determined by LVEDV (0.30 ± 0.05 ml/100 g BW in diabetic versus 0.12 ± 0.02 ml/100 g BW in controls; n = 8, p < 0.05; Figure 1(B)). Treatment with atorvastatin, however, did not modify LVEDV, indicating that whereas the drug improved cardiac function, it did not effectively prevent ventricular dilatation.
Effect of atorvastatin (ATV) on cardiovascular parameters evaluated by echocardiography in control and diabetic rats.

Results are expressed as the mean ± standard error of the mean. n = average of eight rats per group.
p < 0.05 when compared with treated and untreated nondiabetic controls.
p < 0.05 when compared with untreated diabetic rats.

(A) Effect of 4 weeks of treatment with atorvastatin (10 mg/kg/day) on left ventricular end systolic volume (LVESV, ml/100 g body weight [BW]) in diabetic rats and controls. (B) Effect of 4 weeks of treatment with atorvastatin (10 mg/kg/day) on left ventricular end diastolic volume (LVEDV, ml/100 g BW) in diabetic and control rats. The results represent the mean ± standard error of the mean of eight animals per group. * Represents a statistically significant difference (p < 0.05) between diabetic rats and controls. † Represents a statistically significant difference (p < 0.05) between untreated and treated diabetic rats. NS: no statistical significance.
Figure 2 shows the results from experiments in which SBP was determined by cuff sphygmomanometry on the tails of the rats. SBP was higher in untreated diabetic rats than in controls (116.53 ± 3.81 mmHg versus 82.72 ± 2.8 mm Hg, respectively; n = 10, p < 0.05). Daily treatment of diabetic rats with atorvastatin, however, decreased blood pressure to 100.9 ± 5.15 mmHg (n = 10, p < 0.05). In contrast, no effect on SBP was observed in controls after treatment with this HMG-CoA inhibitor.

Effect of 4 weeks of treatment with atorvastatin (10 mg/kg/day) on systolic blood pressure (mmHg) in diabetic rats and controls. The values shown are the means ± standard error of the mean of 10 animals per group. Atorvastatin significantly decreased blood pressure in diabetic rats. * Represents a statistically significant difference (p < 0.05) between diabetic rats and controls. † Represents a statistically significant difference (p < 0.05) between untreated and treated diabetic rats.
Plasma levels of total cholesterol were higher in diabetic rats (241.49 ± 19.89 mg/dl) than in controls (158.08 ± 10.81 mg/dl; n = 5, p < 0.05), and atorvastatin did not reduce these levels in either group (Figure 3). The lack of effectiveness of atorvastatin in reducing cholesterol may indicate that either the current dose of 10 mg/kg/day or the treatment period, or both, needed to be increased.

Effect of 4 weeks of treatment with atorvastatin (10 mg/kg/day) on total cholesterol levels in diabetic rats and controls. Total cholesterol was higher in diabetic rats than in controls, but at this concentration, atorvastatin did not decrease cholesterol levels in diabetic rats. The values shown are the means ± standard error of the mean of five animals per group. * Represents a statistically significant difference (p < 0.05) between untreated diabetic rats and controls. NS: no statistical significance.
Figure 4 depicts the effect of atorvastatin on acetylcholine-induced relaxation. The maximal relaxation (EMAX) achieved in aortic rings with 100 µM acetylcholine was lower in diabetic rats than in controls (53.7 ± 4.1% versus 74.6 ± 3.3%; n = 10, p < 0.05). Atorvastatin restored endothelial function and increased EMAX to 82.1 ± 7.0% (n = 10, p < 0.05) in diabetic rats. EC50 values were similar in diabetic rats (0.56 ± 0.10 µM) and controls (0.41 ± 0.32 µM), and did not change with atorvastatin (0.83 ± 0.32 µM), indicating that the drug does not modify the affinity of the muscarinic receptor for acetylcholine. Note that the addition of 1 mM L-NAME to the incubation bath inhibited acetylcholine-induced relaxation, suggesting that the increased vascular relaxation with atorvastatin is NO-dependent.

Cumulative concentration-response curves for the acetylcholine-induced relaxation of aortic rings from diabetic rats after a 4-week treatment period with atorvastatin (10 mg/kg/day). Note that the addition of 1 mM L-NAME to the incubation bath inhibited the acetylcholine-induced relaxation of aortic rings from treated diabetic rats. Aortic rings were precontracted with 0.1 µM norepinephrine before the addition of cumulative concentrations of acetylcholine. The values shown are the means ± standard error of the mean of 10 animals per group. * Represents a statistically significant difference (p < 0.05) for EMAX between untreated and treated diabetic rats.
Figure 5(A) and (B) show the results from histological studies performed on comparable sections of the thoracic aorta from untreated and atorvastatin-treated diabetic rats and controls. Perivascular fibrosis was higher in diabetic rats than in controls (10.6 ± 0.4 µm/100 g BW in diabetic versus 4.2 ± 0.2 µm/100 g BW in controls; n = 5, p < 0.05). Atorvastatin decreased this variable in diabetic animals from 10.6 ± 0.4 to 8.9 ± 0.4 µm/100 g BW (n = 5, p < 0.05). In addition, the media (figure not shown) was significantly thicker in diabetic rats than in controls, and atorvastatin reduced this parameter in diabetic rats from 49.0 ± 1.0 to 45.0 ± 1.0 µm/100 g BW (n = 5, p < 0.05).
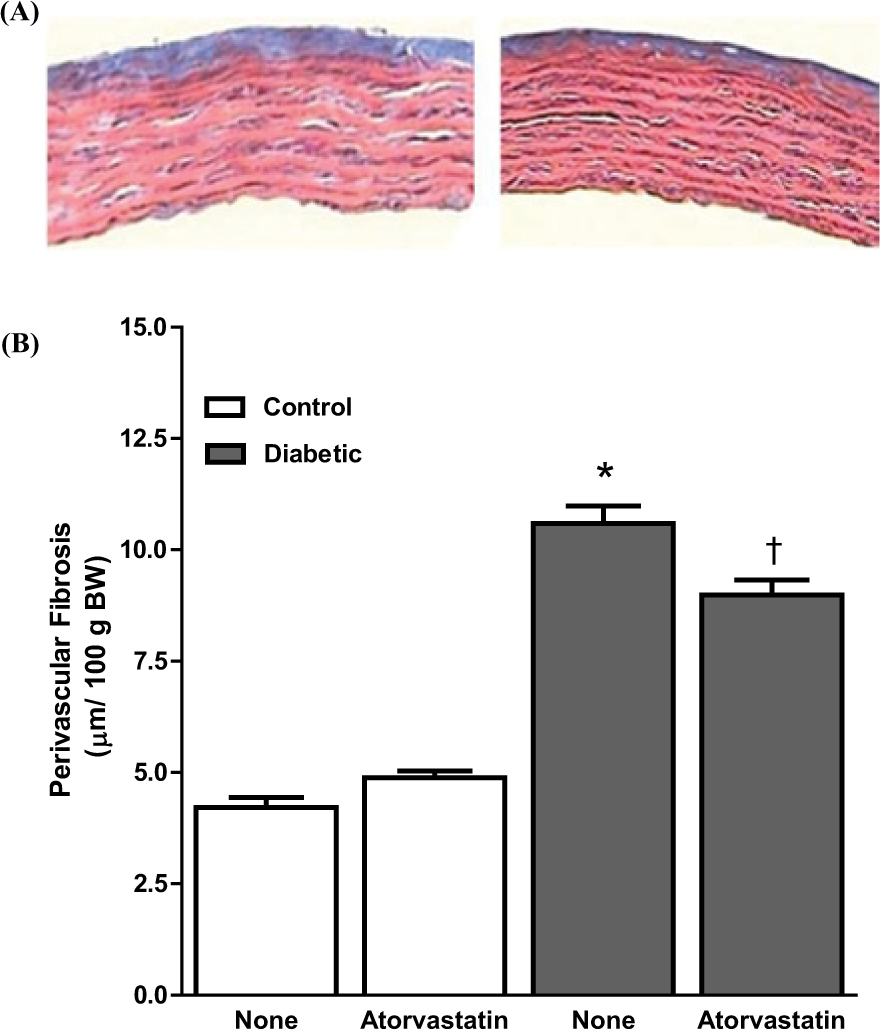
(A) Representative histological sections (×40, Azan–Mallory stain) of aortic segments from untreated (left) and atorvastatin-treated (right) diabetic rats, demonstrating the typical reduction in perivascular fibrosis after treatment with the statin. (B) Quantified thickness of the perivascular fibrosis in comparable aortic segments from atorvastatin-treated and untreated diabetic rats and controls. The mean value of five measurements was reported for each animal. The values shown are the means ± standard error of the mean of five animals per group. * Represents a statistically significant difference (p < 0.05) between untreated diabetic rats and controls. † Represents a statistically significant difference (p < 0.05) between untreated and treated diabetic rats.
Lipid peroxidation was evaluated as an indicator of oxidative stress by measuring cardiac and aortic MDA+4-HAE levels. MDA+4-HAE levels (µmol/g protein) were similar in cardiac homogenates from untreated diabetic rats (1.42 ± 0.12) and controls (1.10 ± 0.12; n = 8, p > 0.05; Figure 6(A)). Atorvastatin did not modify these values. By contrast, in aortic homogenates MDA+4-HAE levels (µmol/g protein) were higher in diabetic rats (6.49 ± 1.24) than in controls (3.69 ± 0.58; n = 8, p < 0.05; Figure 6(B)). Atorvastatin reduced MDA+4-HAE levels in diabetic rats to 2.69 ± 0.42 µmol/g protein (n = 8, p < 0.05).

(A) Effect of atorvastatin (10 mg/kg/day) on MDA+4-HAE levels in cardiac homogenates from diabetic rats and controls. (B) Effect of atorvastatin (10 mg/kg/day) on MDA+4-HAE levels in aortic homogenates from diabetic rats and controls. Atorvastatin reduced lipid peroxidation levels in aortic homogenates from diabetic rats, without affecting these levels in controls. The values shown are the means ± standard error of the mean of eight animals per group. * Represents a statistically significant difference (p < 0.05) between untreated diabetic and control rats. † Represents a statistically significant difference (p < 0.05) between untreated and treated diabetic rats.
Discussion
In the present study, we found that atorvastatin reduces vascular oxidative stress and remodeling, and improves endothelial and CV function in type 1 diabetic rats. At the administered dose of 10 mg/kg/day, however, it does not lower cholesterol and it fails to prevent the development of ventricular dilatation.
Although improved endothelial function has been reported for higher doses of atorvastatin than the 10mg/kg/day dose that was used in this study, we found that it was effective in normalizing endothelial function. A 20 mg/kg/day dose improves endothelial function in spontaneously hypertensive rats [Wassmann et al. 2001], and a 50 mg/kg/day dose improves endothelium-dependent vasodilatation in STZ diabetic rats [Riad et al. 2007]. A 40 mg/day dose improves endothelial function in type 1 diabetic patients after administration over a 6-week period [Dogra et al. 2005], and in diabetic patients without coronary heart disease or hypertension over a 6-month period [Konduracka et al. 2008]. Controversy still exists on this topic, however. Neither a 40 mg/day dose nor an 80 mg/day dose of atorvastatin prevents coronary endothelial dysfunction in swine with left ventricular hypertrophy induced by aortic banding [Forcillo et al. 2011]. In addition, atorvastatin does not improve endothelial function in type 2 diabetic patients with average cholesterol levels and without prior CV disease [Tantikosoom et al. 2005]. Moreover, supplementing the diet of diabetic rats with simvastatin does not improve endothelial-dependent relaxation in the mesenteric arteries [Palmer et al. 1998]. Differences in experimental animal models, patient comorbidities, statin doses, and treatment duration may partly explain these diverse findings.
The mechanisms underlying vascular protection provided by statins has yet to be established. That the improved endothelial function is fully abolished by L-NAME and is independent of cholesterol levels, suggests that atorvastatin improves vascular function by increasing NO availability. This action may result from NOS activation, NO scavenger reduction, or a combination of both factors. Reductions in lipid peroxidation in aortic homogenates suggest that at low doses, atorvastatin has antioxidant properties. At higher doses, atorvastatin decreases eNOS uncoupling and superoxide production in diabetic rats [Wenzel et al. 2008], and reduces reactive oxygen species (ROS) in rat aortic segments [Wassmann et al. 2001, 2002]. It also upregulates eNOS and nNOS expression in the kidney of spontaneously hypertensive rats [Ito et al. 2010]. Similarly, simvastatin and lovastatin increase eNOS mRNA and augment eNOS function in endothelial cells from human saphenous vein [Laufs et al. 1998]. These findings suggest that the antioxidant effects found with atorvastatin may be class-related. It should be noted, however, that Elmadhun and colleagues report that atorvastatin increases myocardial and serum oxidative stress in a porcine model for metabolic syndrome [Elmadhun et al. 2012].
Previously, we demonstrated that cardiac function is compromised in STZ diabetic rats at four weeks following diabetes induction [Crespo et al. 2003, 2011]. At this stage, dilated cardiomyopathy is measurable by echocardiography [Crespo et al. 2003]. In the current study, we reveal that a low dose of atorvastatin administered over a 4-week period increases ejection fraction, stroke volume, and CO, and significantly reduces end-systolic volume. End-diastolic volume, however, is not reduced, suggesting that, even though atorvastatin improves systolic function, it does not improve diastolic function or prevent ventricular dilatation. In line with our findings, Chen and colleagues reported that a higher dose of atorvastatin (100 mg/kg/day) was not effective in preventing the onset of diastolic dysfunction in a type 2 diabetic rat model [Chen et al. 2007]. Furthermore, similar to our findings on systolic performance, clinical trials show that atorvastatin improves left ventricular systolic function and symptoms of heart failure in patients with nonischemic cardiomyopathy [Sola et al. 2006] and chronic heart failure [Yamada et al. 2007; Xie et al. 2010]. However, not all statins are equally effective in improving systolic function. Rosuvastatin, for example, does not have a positive impact on clinical outcomes in patients with chronic heart failure and does not reduce the number of deaths in older patients with systolic heart failure [Tavazzi et al. 2008; Lipinski et al. 2009].
The mechanisms underlying the ability of atorvastatin to improve systolic function are still undefined. This study reveals that both LVESV and ejection fraction improve after treatment with this statin, suggesting that improved systolic function may be secondary to a decrease in peripheral resistance associated with improved endothelial function, reduced SBP, and regression of vascular remodeling with atorvastatin treatment. Reduced peripheral resistance may increase cardiac function by imposing less work on the myocardium. Alternatively, atorvastatin may preserve the contractile elements in myocardial tissue, which are deteriorated in diabetes. Indeed, 5 days of treatment with simvastatin (10 mg/kg/day) improves contractile function in isolated hearts from diabetic hypercholesterolemic rats, independently of the cholesterol-lowering effects of this drug [Adameova et al. 2009]. Additional experiments are needed to confirm this alternative.
It is important to note that in addition to decreasing cholesterol levels, statins decrease coenzyme Q10 synthesis [Laaksonen et al. 1994]. Both cholesterol and coenzyme Q10 share the same biosynthetic pathway. This coenzyme is essential for the production of ATP in the mitochondrial electron transport system [Crane, 2001], and a decrease in Q10 levels has been associated with mitochondrial dysfunction and myopathies [De Pinieux et al. 1996]. Indeed, results from a pilot study indicate that concomitant administration of coenzyme Q10 to hypercholesterolemic patients reduces muscle pain associated with statin treatment [Caso et al. 2007]. Although Q10 deficiency may affect energy metabolism in cardiac muscle, a direct effect of statins has not been reported in this tissue. The effect of atorvastatin on Q10 levels is not clear. On the one hand, neither 20 mg pravastatin nor 10 mg atorvastatin was found to decrease Q10 level in healthy volunteers despite a significant reduction in cholesterol concentration [Bleske et al. 2001]. On the other hand, atorvastatin significantly reduced Q10 concentration in patients at risk for CV disease [Rundek et al. 2004]. Regardless of the potential reduction of Q10 levels by atorvastatin, the results from the current work suggest that its benefits, particularly in reducing vascular oxidative stress and remodeling, and in improving CV function, make this statin a potential agent for reducing CV complications in diabetes.
If the current study with type 1 diabetic rats is applicable to humans, diabetic patients who are prone to develop CV complications, including impaired systolic performance, may gain benefits from atorvastatin that extend beyond the reduction of cholesterol levels. This is particularly relevant for type 1 diabetic patients. Poor glycemic control has been established as prognostic of a higher mortality rate in patients with acute myocardial infarction [Kosiborod et al. 2009], and a powerful association has been established between elevated glucose levels and the risk of death among patients with acute heart failure [Mebazaa et al. 2013]. Moreover, in a cohort of the Epidemiology of Diabetes Interventions and Complications (EDIC) study, alterations in cardiac function and remodeling in type 1 diabetic patients were associated with prior glycemic exposure [Genuth et al. 2013]. Thus, although atorvastatin does not have any effect on reducing glucose levels, its ability to reduce vascular oxidative stress and remodeling and to improve CV function suggests that concomitant use of statins and hypoglycemic agents can prove useful in reducing CV complications for both type 1 diabetics and those patients with poor glycemic control. Additional studies addressing the effect of different HMG-CoA reductase inhibitors on both diabetic patients and animal models of diabetes are needed to determine the optimal statin regimen, should it be used as an adjuvant to conventional glycemic therapies.
Footnotes
Acknowledgements
The authors would like to recognize the assistance of Dr Donald C. Dunbar for editing the manuscript.
Funding
This work was supported by the National Institute of Health (MBRS-RISE Grant R25GM061838, RCMI Grant G12-RR03051) and the Associated Deanship of Biomedical Sciences and Graduate Studies of the UPR School of Medicine.
Conflict of interest statement
The authors have no conflicts of interest to declare with respect to the authorship and/or publication of this article.