
Abstract
Although low-density lipoprotein cholesterol (LDL-C) lowering represents the mainstay of current lipid treatment, high-density lipoprotein cholesterol (HDL-C) has generated increasing interest as a secondary therapeutic target because of strong evidence that serum HDL-C concentration is inversely associated with coronary heart disease risk. Niacin is a lipid-altering drug that has been used to lower cholesterol since the 1950s. In addition to its LDL-C-lowering effects, niacin is the most effective agent currently available for raising HDL-C. Despite its long history as a lipid-altering drug, only limited data are available regarding its clinical benefit alone and in combination with other agents, and the majority of studies investigating its impact on clinical outcomes are from the pre-statin area. Several studies have demonstrated a beneficial effect of treatment with niacin in combination with statin therapy on surrogate cardiovascular markers (e.g. carotid intima-media thickness). However, the clinical significance of these surrogate markers has been questioned. Two large randomized trials will address whether niacin–statin combination therapy is an appropriate therapeutic alternative to statin monotherapy.
Introduction
Dyslipidemia is a well established risk factor for cardiovascular disease [Sniderman et al. 2011; Lewington et al. 2007; Wilson et al. 1998; Stamler et al. 1986; Lipid Research Clinics Program, 1984]. Multiple clinical trials of lipid-lowering agents have demonstrated that lowering low-density lipoprotein cholesterol (LDL-C) effectively reduces the risk of cardiovascular events and death in patients with and without known coronary heart disease (CHD) [Baigent et al. 2010; Downs et al. 1998; Lipid Study Group, 1998; Shepherd et al. 1995; Lipid Research Clinics Program, 1984]. As a result, lowering LDL-C is an important therapeutic target for both primary and secondary prevention of cardiovascular events.
Current treatment guidelines identify LDL-C lowering as the primary goal of therapy [Genest et al. 2009; Graham et al. 2007; National Cholesterol Education Program, 2001]. These guidelines focus on treatment with 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins), given their superior efficacy relative to other LDL-C-lowering agents [Genest et al. 2009; Graham et al. 2007; National Cholesterol Education Program, 2001]. Not all patients on statin monotherapy achieve target LDL-C levels, however, and even when LDL-C lowering is successful there remains significant residual cardiovascular risk [Cannon et al. 2004; Heart Protection Study Collaborative Group, 2002; Lipid Study Group, 1998; Shepherd et al. 1995; Scandinavian Simvastatin Survival Study Group, 1994]. Although efficacy is improved with higher statin doses [Baigent et al. 2010], high-dose therapy carries a higher risk of side effects such as myopathy [Armitage et al. 2010]. For these reasons, there is increasing interest in developing combination LDL-C-lowering therapies that may augment the treatment effect and minimize the side effects of statins.
While LDL-C lowering represents the mainstay of current lipid treatment, high-density lipoprotein cholesterol (HDL-C) has generated increasing interest as a secondary therapeutic target [National Cholesterol Education Program, 2001]. There is strong evidence that serum HDL-C concentration is inversely associated with CHD risk, which has led to the hypothesis that HDL-C protects against atherosclerosis [Vergeer et al. 2010]. Experimental data have led to the identification of several mechanisms for this potential atheroprotective role, and clinical studies have suggested that raising HDL-C may reduce cardiovascular events [Natarajan et al. 2010]. However, since these benefits have largely been seen in the setting of multiple lipid changes, it has been difficult to isolate the specific effects of raising HDL-C. Although conclusive evidence demonstrating the value of targeting HDL-C is lacking, some of the investigative interest in combination therapies has centered on agents that specifically raise HDL-C.
Comparison of different drugs affecting lipid metabolism.

Not approved by FDA.
CETP, cholesterol ester transfer protein; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; PCSK9, proprotein convertase subtilisin/kexin type 9.
Mechanism of action
The lipid-modifying effects of niacin were first reported in 1955, when it was noted that niacin decreased total cholesterol and LDL-C and increased HDL-C [Altschul et al. 1955]. More recent studies have confirmed these initial findings, and have also shown a differentially dose-dependent effect of niacin on LDL-C and HDL-C [Guyton et al. 2000. 1998; Luria, 1988; Vega and Grundy, 1994]. Whereas niacin-induced changes in HDL-C are logarithmic, changes in LDL-C are linear [Illingworth et al. 1994]. As a result, relatively small doses of niacin (1.5–2.0 g/day) produce substantial increases in HDL-C (approximately 15–40%), making it the most effective lipid-altering drug for raising HDL-C. By contrast, doses of at least 3.0–4.5 g/day may be necessary to achieve LDL-C reductions of approximately 15% [Knopp, 1999]. In addition to its LDL-C- and HDL-C-altering effects, niacin has been shown to decrease serum triglycerides by 20–50% and lipoprotein(a) by about 20% [Carlson et al. 1989]. The effect on triglycerides appears to be clinically beneficial, given the growing evidence implicating triglyceride-rich lipoproteins in atherogenesis, but the significance of lipoprotein(a) reduction remains unclear [National Cholesterol Education Program, 2001].
Niacin lowers serum LDL-C through several mechanisms (Figure 1). It inhibits the peripheral mobilization of free fatty acids [Wu and Zhao, 2009], which decreases the substrate available for hepatic synthesis of triglycerides and very low-density lipoprotein (VLDL) particles [Grundy et al. 1981]. This in turn reduces hepatic conversion of VLDL particles to LDL particles. In addition, niacin appears to interfere directly with the enzymatic process that mediates the conversion of VLDL-C to LDL-C [Grundy et al. 1981] and decreases triglyceride synthesis and hepatic lipoprotein secretion via inhibition of diacylglycerol acyltransferase 2 [Ganji et al. 2004].
Mechanisms of action of niacin. apoA-I, apolipoprotein A-I; CETP, cholesteryl ester transfer protein; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; VLDL-C, very low-density lipoprotein cholesterol.
Niacin’s effect on HDL-C levels is mediated in part by the reduction in LDL-C and VLDL-C levels, since reduced availability of these lipoproteins limits cholesterol transfer from HDL-C to LDL-C and VLDL-C, thereby increasing serum HDL-C levels [Natarajan et al. 2010]. However, this mechanism cannot completely account for niacin’s substantial effect on HDL-C levels. Studies in both mouse and human models have identified several other novel mechanisms that act in parallel [Natarajan et al. 2010]. First, niacin decreases the concentration and activity of cholesteryl ester transfer protein (CETP), an enzyme that transfers cholesterol from HDL particles to LDL and VLDL particles. The result is to preserve HDL particles at the expense of LDL and VLDL particles [Van Der Hoorn et al. 2008]. Second, niacin selectively decreases hepatic excretion of apolipoprotein A-I, the major protein component of HDL-C in plasma, probably by inhibiting the hepatocyte surface expression of β-chain adenosine triphosphate synthase, a recently reported HDL–apolipoprotein A-I holoparticle receptor [Martinez et al. 2003]. This allows more apolipoprotein A-I-containing HDL-C to re-enter the circulation from the liver [Jin et al. 1997]. In addition to raising HDL-C levels, niacin appears to act directly on atherosclerotic plaques to promote HDL-mediated cholesterol efflux from arterial wall macrophages, a process known as reverse cholesterol transport [Al-Mohaissen et al. 2010; Zambon et al. 1999].
Side effects
Although the lipid-altering benefits of niacin are widely recognized, there are several well known side effects that limit its use. Flushing of the skin, which is caused by prostaglandin-mediated vasodilation [Al-Mohaissen et al. 2010], is the most frequent side effect, occurring in at least 80% of patients taking standard crystalline preparations [Gibbons et al. 1995]. While some studies suggest that the flushing response is actually associated with improved response to therapy [Knopp, 1999], it is an intolerable side effect for some people and was a common reason for medication discontinuation in earlier generations of the drug. This reaction can be minimized, however, by administration of aspirin prior to each dose or by taking the drug at the end of meals [Knopp, 1999]. In addition, a once daily extended-release formulation is now available that causes less frequent and less severe flushing than standard crystalline formulations (approximately 20% compared with 40% discontinuation rate due to flushing) [Maccubbin et al. 2009; Gibbons et al. 1995]. Moreover, selective prostaglandin-2 receptor inhibitors, such as laropiprant, have been developed and formulated with niacin to further reduce flushing (approximately 10% compared with 20% discontinuation rate due to flushing) [Hussein et al. 2010; Maccubbin et al. 2009].
Other adverse effects of niacin include hepatotoxicity, hyperuricemia, and hyperglycemia. Hepatotoxicity was of particular concern in earlier generation sustained-release formulations but appears to be fairly uncommon with extended-release niacin (significant hepatic transaminase elevations occur in <1% of patients) [Capuzzi et al. 1998]. There are reports of myopathy in patients treated with niacin and a statin [Guyton and Bays, 2007]; however, the risk of rhabdomyolysis with niacin remains unclear. Previous studies had not suggested a general myopathic effect of niacin. However, the US Food and Drug Administration (FDA) has refocused attention on the issue of myopathy related to lipid therapies and recently issued a warning for high-dose simvastatin (80 mg) because of increased risk of muscle damage [US Food and Drug Administration, 2011]. Results from two large randomized controlled trials will help clarify the risk of rhabdomyolysis with niacin [Aim-High Investigators, 2011].
The effect of niacin on serum glucose regulation remains a significant concern, although the clinical significance of this effect remains somewhat controversial. The Assessment of Diabetes Control and Evaluation of the Efficacy of Niaspan® Trial (ADVENT), which randomly assigned people with type 2 diabetes to niacin or placebo, demonstrated a small but significant worsening in glycemic control in one of the niacin arms [Grundy et al. 2002]. Specifically, while the baseline and week 16 values for glycosylated hemoglobin levels remained unchanged at 7.1% in the placebo group, they increased from 7.2% (baseline) to 7.5% (week 16) in the extended-release niacin group dosed at 1.5 g/day. No difference was observed in the group treated with 1.0 g/day of extended-release niacin. Because hyperglycemia appears to be dose-dependent, lower doses of niacin (<2.0 g/day) are recommended for diabetic patients [National Cholesterol Education Program, 2001].
Early clinical studies with cardiovascular endpoints
Early clinical studies with cardiovascular endpoints.

All-cause mortality relative risk reduction 11% (p = 0.0004) at 9-year follow up.
poB, apolipoprotein B; CHD, coronary heart disease; CI, confidence interval; CV, cardiovascular; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; TIA, transitory ischemic attack.
Although the findings of the CDP provide the strongest evidence for the clinical benefit of niacin to date, the trial was conducted in an era when many other therapies that have subsequently been shown to reduce recurrent ischemic events were either not widely used (e.g. aspirin and β-blockers) or not available (e.g. statins, thienopyridines, and angiotensin-converting enzyme inhibitors). Thus, the clinical significance of findings from studies conducted over three decades ago is limited.
To address this shortcoming of prior studies, three other clinical trials evaluated the cardiovascular outcomes of niacin treatment in combination with other lipid-modifying therapies, namely fibrates in the Stockholm Ischaemic Heart Disease Secondary Prevention Study [Carlson and Rosenhamer, 1988], bile acid sequestrants and statins in the Familial Atherosclerosis Treatment Study (FATS) [Brown et al, 1998, 1990], and statins in the HDL-C-Atherosclerosis Treatment Study (HATS) [Brown et al. 2001].
In the Stockholm Ischaemic Heart Disease Secondary Prevention Study, 555 patients post-MI were randomized to open-label niacin (3.0 g/day) in combination with clofibrate (2.0 g/day) or placebo and followed for a total of 5 years [Carlson et al. 1988]. At the end of the trial period, patients in the treatment group had a 26% relative risk reduction in all-cause mortality (21.9% versus 29.7%; p < 0.05) and a 36% relative risk reduction in cardiovascular mortality (16.8% versus 26.4%; p < 0.01) relative to the control group. Because of the trial design, however, it is not possible to determine how much of the benefit is due to niacin, clofibrate, or their combination. Interestingly, the treatment effect was most pronounced in patients with reductions in serum triglyceride levels of greater than 30%, but the benefit did not correlate with changes in serum total cholesterol. The lack of association with changes in total serum cholesterol may have been related to a low prevalence of hypercholesterolemia in the study population as well as to niacin’s opposite effects on LDL-C and HDL-C, which are not captured by measuring total cholesterol.
The FATS study was designed to assess the effect of intensive lipid-lowering therapy on the progression of coronary atherosclerosis [Brown et al. 1990]. A total of 146 men with elevated apolipoprotein B levels, documented CHD, and a family history of vascular disease were randomized to either niacin (4.0–6.0 g/day) plus the bile acid sequestrant colestipol (30 g/day), lovastatin (40 mg/day) plus colestipol (30 mg/day), or ‘conventional therapy’ (placebo or colestipol monotherapy if LDL-C was elevated). After 2.5 years, the average degree of coronary stenosis as measured by quantitative arteriography improved in the niacin plus colestipol group and in the lovastatin plus colestipol group (change in coronary diameter +0.9% and +0.7%, respectively) and worsened with conventional therapy (−2.1%) (p = 0.003 for niacin plus colestipol compared with placebo). In addition, the rates of cardiovascular events (death, myocardial infarction, and refractory ischemic symptoms requiring coronary or peripheral revascularization) in patients treated with niacin plus colestipol and with lovastatin plus colestipol were 4.2% and 6.5%, respectively, compared with a rate of 19.2% in the conventional therapy group. This represented a 73% relative risk reduction (95% confidence interval 23–90%) in cardiovascular clinical events associated with intensive lipid-lowering therapy [Brown et al. 1990]. A 10-year post hoc follow-up study of FATS that compared patients who were treated with aggressive open-label triple-drug therapy (niacin 4.0–6.0 g/day plus colestipol 30 g/day plus lovastatin 40 mg/day) with conventional therapy following the formal trial period demonstrated a 67% relative risk reduction in cardiovascular events (5.3% compared with 18.8%; p < 0.02) and a 93% relative risk reduction inall-cause mortality (1.3% compared with 19.8%; p < 0.001) in patients receiving triple-drug therapy [Brown, 2006]. Although not a formally randomized comparison, the results of this follow-up study suggested a long-term clinical benefit of combining niacin with other lipid-lowering agents. Again, the trial design, which combined various lipid agents, makes it difficult to know which therapies contributed to the benefit.
Most recently, the HATS trial was a double-blind, randomized, placebo-controlled study that evaluated extended-release niacin (2.0–4.0 g/day) in combination with simvastatin (10–20 g/day) in comparison with placebo in 160 patients with CHD, HDL-C ≤35 mg/dl in men or ≤40 mg/dl in women, and LDL-C ≤145 mg/dl [Brown et al. 2001]. As in the FATS trial, both angiographic and clinical endpoints were used. The study showed a slight regression in coronary stenosis in the niacin plus simvastatin treatment arm compared with placebo (change in coronary diameter −0.4% compared with +3.9%; p < 0.001) [Brown et al. 2001]. In addition, the composite cardiovascular clinical endpoint of death, MI, stroke, or revascularization was significantly reduced by 90% in patients treated with niacin plus simvastatin compared with placebo (2.6% compared with 23.7%; p = 0.03) [Brown et al. 2001]. The authors concluded that the addition of niacin to simvastatin therapy in CHD patients with low HDL-C and ‘normal’ LDL-C resulted in slight regression of coronary atherosclerosis and a substantial reduction in clinical coronary events over 3 years. Because there was no statin monotherapy arm of the trial, however, it is impossible to determine the relative contributions of niacin and simvastatin to the observed clinical benefit. Nonetheless, HATS laid the foundation for the contemporary investigative interest in adding niacin to statin therapy.
Clinical studies with surrogate endpoints
Clinical studies with surrogate endpoints.
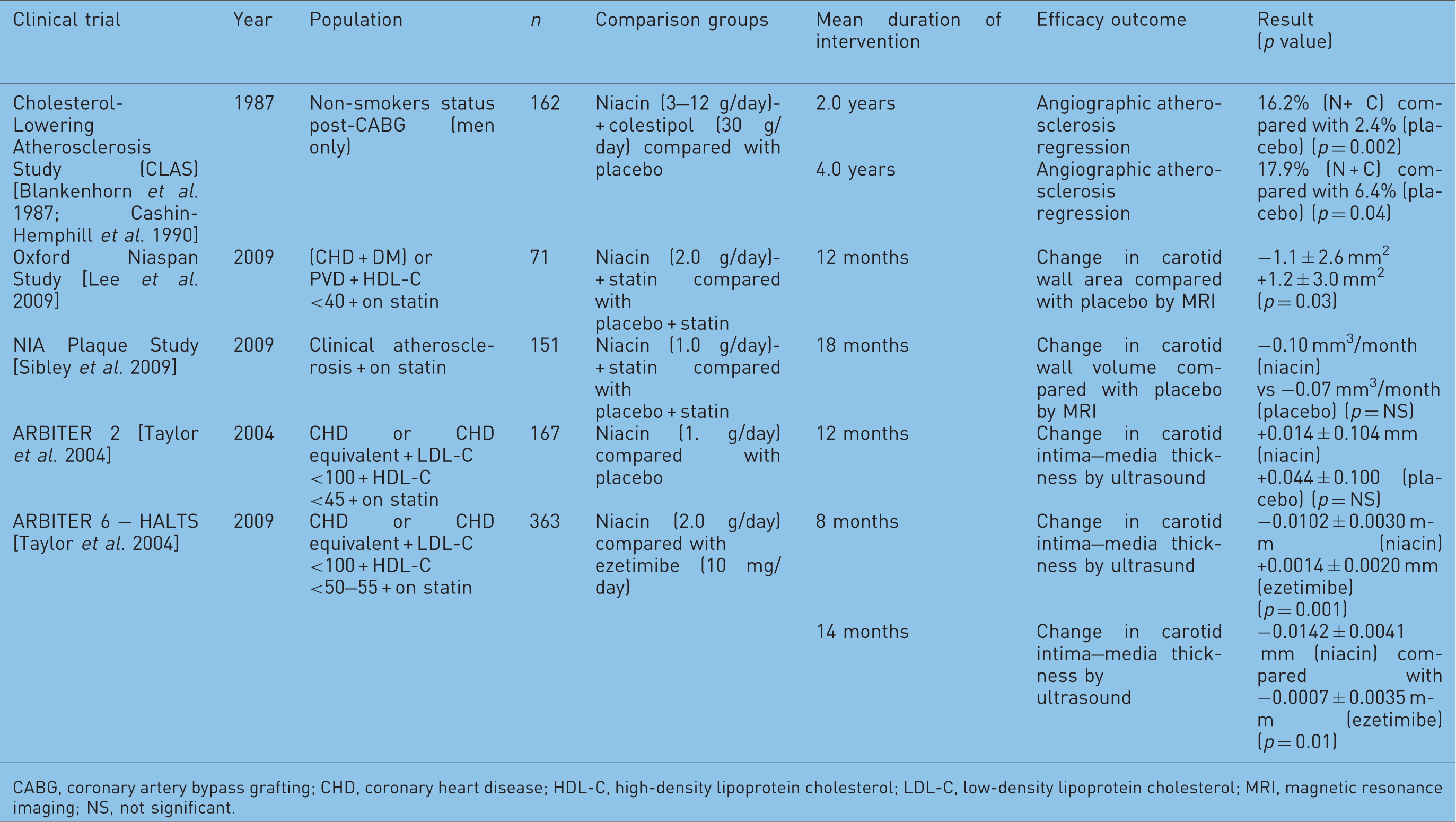
CABG, coronary artery bypass grafting; CHD, coronary heart disease; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; MRI, magnetic resonance imaging; NS, not significant.
Since then, investigators have explored noninvasive imaging modalities as biomarkers for cardiovascular disease risk. Two of these studies, the Oxford Niaspan Study and the NIA Plaque Study, sought to evaluate niacin’s effects on carotid atherosclerosis using carotid magnetic resonance imaging (MRI). In the Oxford Niaspan Study, 71 patients with low HDL-C (<40 mg/dl) and either type 2 diabetes with CHD or carotid/peripheral atherosclerosis were randomized in a double-blind fashion to extended-release niacin (2.0 g/day) plus statin therapy (the specific agent and dose were determined by the physician) or placebo plus statin therapy. After 1 year, the extended-release niacin group had a significant reduction in mean carotid wall area compared with placebo (−1.1 ± 2.6 mm2 compared with + 1.2 ± 3.0 mm2; p = 0.03) [Lee et al. 2009]. In the NIA Plaque Study, 151 patients were randomized to extended-release niacin (1.5 g/day) or placebo in addition to background statin therapy and were followed for 18 months. Although a significant reduction in LDL-C andasignificant increase in HDL-C were observed in the extended-release niacin group, no significant difference in plaque regression was noted between the two groups (change in MRI-measured wall volume was −0.10 mm3/month in the niacin group compared with −0.07 mm3/month in the placebo group) [p=NS] [Sibley et al. 2009].
The Arterial Biology for the Investigation of Treatment Effects of Reducing Cholesterol (ARBITER) trials were also designed to assess the value of adding extended-release niacin to statin therapy, but the investigators used change in carotid intima–media thickness by ultrasonography, a validated predictor of cardiovascular events [O’Leary et al. 1999], as the primary outcome measure. The primary objective of ARBITER 2, a double-blind, randomized, placebo-controlled trial, was to determine theeffect of extended-release niacin (1.0 g/day) in comparison with placebo on carotid intima–media thickness when added to background statin therapy in patients with known CHD and good LDL-C control (<130 mg/dl) but low HDL-C levels (<45 mg/dl). Although the change in carotid intima–media thickness observed in the niacin group relative to placebo was not significant (+0.014 ± 0.104 mm compared with +0.044 ± 0.100 mm, p = 0.08), the trend suggested the potential for statin–niacin combination therapy to decrease carotid atherosclerosis and possibly improve cardiovascular outcomes [Taylor et al. 2004].
The ARBITER 6–HALTS trial compared the effects of HDL-C and LDL-C-focused lipid modification strategies on the same surrogate endpoint of carotid intima–media thickness. Patients with CHD or a CHD risk equivalent, already on long-term statin therapy, with optimal LDL-C control (<100 mg/dl) and borderline HDL-C (<50 mg/dl for men and <55 mg/dl for women) were randomized to extended-release niacin with a target dose of 2.0 g/day (HDL-C-focused strategy) or ezetimibe at 10 mg/day (LDL-C-focused strategy). The trial was stopped early, as patients in the niacin group had a significant reduction in carotid intima–media thickness relative to those in the ezetimibe group at 8 months (−0.0102 ± 0.0030 mm compared with +0.0014 ± 0.0020 mm; p = 0.001) and 14months of follow up (−0.0142 ± 0.0041 mm compared with −0.0007 ± 0.0035 mm; p = 0.01) [Taylor et al. 2009].
Although the ARBITER 6–HALTS trial received considerable attention as evidence favoring an HDL-C-oriented strategy when adding a second agent to an optimal statin regimen, it is difficult to interpret the clinical significance of this study given the use of carotid intima–media thickness as a surrogate endpoint. This point was underscored in a recent meta-analysis of 41 randomized clinical trials designed with carotid intima–media thickness regression as a biomarker for reduction in cardiovascular events [Costanzo et al. 2010]. Using a weighted random effects meta-regression analysis, the authors tested the relationship between mean and maximum carotid intima–media thickness changes and outcomes. In contrast to earlier evidence suggesting the utility of carotid intima-media thickness as a predictor of cardiovascular events, the analysis revealed no significant relationship between carotid intima–media thickness regression and cardiac ischemic events, cerebrovascular events, or all-cause mortality. Another analysis showed that only one of the various ways to assess carotid intima-media thickness was an independent but rather modest predictor of cardiovascular risk [Polak et al., 2011]. Thus, it is currently uncertain whether adding niacin to statin therapy meaningfully reduces a patient’s cardiovascular risk.
Large clinical trials in the modern era
In order to address more directly the question of whether patients with cardiovascular disease who have well controlled serum LDL-C levels on a statin but who have persistent atherogenic dyslipidemia can benefit from niacin–statin combination therapy, two large clinical trials were designed (Table 4). The Atherothrombosis Intervention in Metabolic syndrome with low HDL-C/high triglycerides: Impact on Global Health outcomes (AIM-HIGH) trial was a randomized trial that enrolled 3414 subjects from the USA and Canada with established cardiovascular disease and atherogenic dyslipidemia [Aim-High Investigators, 2011] (Figure 2). All patients were treated with simvastatin or simvastatin plus ezetimibe (10 mg/day) to achieve a goal LDL-C in the range of 40–80 mg/dl; in addition, they were randomized to treatment with either extended-release niacin (1.5–2.0 g/day) or placebo. The primary efficacy outcome was time to first cardiovascular event, defined as the composite of CHD death, nonfatal MI, ischemic stroke, hospitalization for acute coronary syndrome, or symptom-driven coronary or cerebral revascularization. The trial completed enrollment in April 2010 and follow up was planned to continue to 2012 [Aim-High Investigators, 2011]. However, the National Heart, Lung, and Blood Institute of the National Institutes of Health (USA) stopped this trial 18 months earlier than planned because of futility (i.e. no clinical benefit was being observed with niacin and continuation of the trial to the end was unlikely to show a difference) [National Heart Lung and Blood Institute, 2011]. The combination of extended-release niacin and a statin had increased HDL-C and lowered triglyceride levels compared with participants who took a statin alone but had not reduced fatal or nonfatal heart attacks, strokes, hospitalizations for acute coronary syndrome, or revascularization procedures. There was even a small increase in ischemic stroke rate in the niacin group (niacin 1.6%, control 0.7%). It remains unclear whether this was due to the play of chance, however, since 9 of the 28 strokes in the niacin group occurred in subjects who had discontinued niacin at least 2 months before their stroke.
AIM-HIGH trial design. CV, cardiovascular; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; TG, triglycerides. Currently ongoing trials with clinical outcomes. ACS, acute coronary syndrome; CV, cardiovascular; CVD, cardiovascular disease; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction.
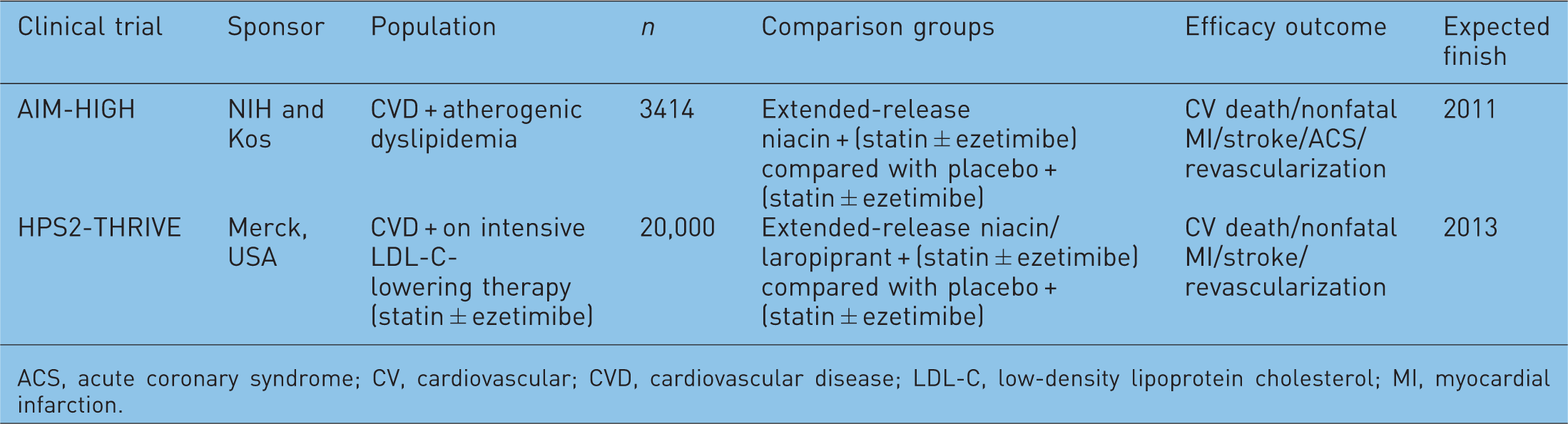
The second trial, Treatment of HDL-C to Reduce the Incidence of Vascular Events (HPS2-THRIVE), is the largest trial to date evaluating the efficacy of niacin therapy. A totalof 25,000 patients with a history of cardiovascular disease (prior myocardial infarction, cerebrovascular disease, peripheral vascular disease, or diabetes with symptomatic CHD) already on intensive LDL-C-lowering therapy with simvastatin 40 mg/day or simvastatin/ezetimibe 40/10 mg/day will be randomized to extended-release niacin (2.0 g/day) plus laropiprant, a selective prostaglandin-2 receptor inhibitor that reduces prostaglandin-mediated flushing, or placebo (Figure 3). The primary efficacy outcome of HPS2-THRIVE is time to first cardiovascular event, a composite of coronary death, nonfatal MI, ischemic stoke, and requirement for arterial revascularization. Secondary outcomes include each of these major adverse cardiovascular events individually. This trial thus seeks to provide direct clinical evidence that therapy directed at raising HDL-C has clinical cardiovascular benefit when used in combination with aggressive LDL-C-lowering therapy in high-risk patients.
HPS2-THRIVE trial design. CV, cardiovascular; MI, myocardial infarction.
Conclusion
Interest in the use of novel lipid-altering combination therapies designed to target atherogenic dyslipidemia has brought niacin, a drug nearly 60 years old, back into the spotlight. Early clinical trial evidence from the CDP demonstrated the clinical benefit of niacin monotherapy, but surprisingly little data are available regarding the utility of niacin in combination with contemporary intensive LDL-C-lowering agents like the statins. Those trials that have explored niacin–statin combinations have been fairly limited in size, have relied on surrogate imaging endpoints that may or may not be good predictors of clinical cardiovascular benefit, and/or were not designed to identify the independent contribution of niacin. Thus, while older trials suggest a potential clinical benefit for niacin, the preliminary results of AIM-HIGH suggest that there is no clinical benefit of niacin added to statin treatment. The ongoing HPS2-THRIVE trial will provide further insights into the lingering question of whether aggressive raising of HDL-C with niacin can improve cardiovascular outcomes beyond what is already achieved with intensive statin monotherapy. Even if the results of HPS2-THRIVE are positive, niacin will need to be compared with treatments that markedly (>50%) increase HDL-C (e.g. CETP inhibitors [Mitka, 2011]) and are evaluated in the context of newer therapies that achieve marked (>60%) reduction in LDL-C [e.g. inhibitors of proprotein convertase subtilisin/kexin type 9 (PCSK9) [Duffet al. 2011]].
Funding
Dr Hochholzer reports research grant support from Roche Diagnostics and honoraria from consulting from Sanofi-Aventis. Dr Giugliano reports research grant support from Amgen, Daiichi Sankyo and Merck, and honoraria from consulting and/or CME lectures from Amgen, Bristol-Myers Squibb, Daiichi Sankyo, and Merck.
Conflict of interest statement
Dr Berg reports no conflicts.