
Abstract

Introduction
Cystic Fibrosis (CF) is the most common autosomal recessive genetic disease in the Caucasian population. It is caused by mutations in the gene Cystic Fibrosis Transmembrane Conductance Regulator (CFTR), with the most frequent mutation being F508del. 1 Respiratory involvement is the primary determinant of the progression of this disease, characterised by impaired mucociliary clearance that leads to recurrent lung infections, persistent chronic inflammation of the airways, development of bronchiectasis and progressive lung damage. 1 Since its first clinical description in 1938, we have witnessed an extraordinary change in the treatment and prognosis of patients with CF. Lessons learned during this process could serve as a reference for other, more common, respiratory diseases. Below we discussed some of these lessons.
Lesson 1: Targeted therapies
CF was traditionally considered a childhood disease with a high risk of early mortality and a life expectancy that did not exceed 18 years. 2 However, the introduction of new drugs, such as inhaled antibiotics and, more recently, CFTR modulators (CFTRm) that functionally correct the genetic defect, along with the establishment of standards for disease management in specialised and multidisciplinary units, have radically changed the life expectancy of people with CF (pwCF). Today, CF has shown one of the most successful improvements in chronic diseases with the greatest increase in survival over the past 40 years. 3 As a result, the number of adults with CF has now surpassed the number of paediatric patients, forcing the health care systems to adapt to this new patient profile. 4
The ageing of the CF population is also accompanied by an increase in the prevalence of comorbidities, which poses a significant burden for the patient and their family on physical, psychological, social and economic levels, from birth to death. 5 All these nuances and needs arising from the complex management of pwCF make CF a pioneer in the research and innovation of new therapeutic and care approaches that have enabled a patient-centred approach (Figure 1).
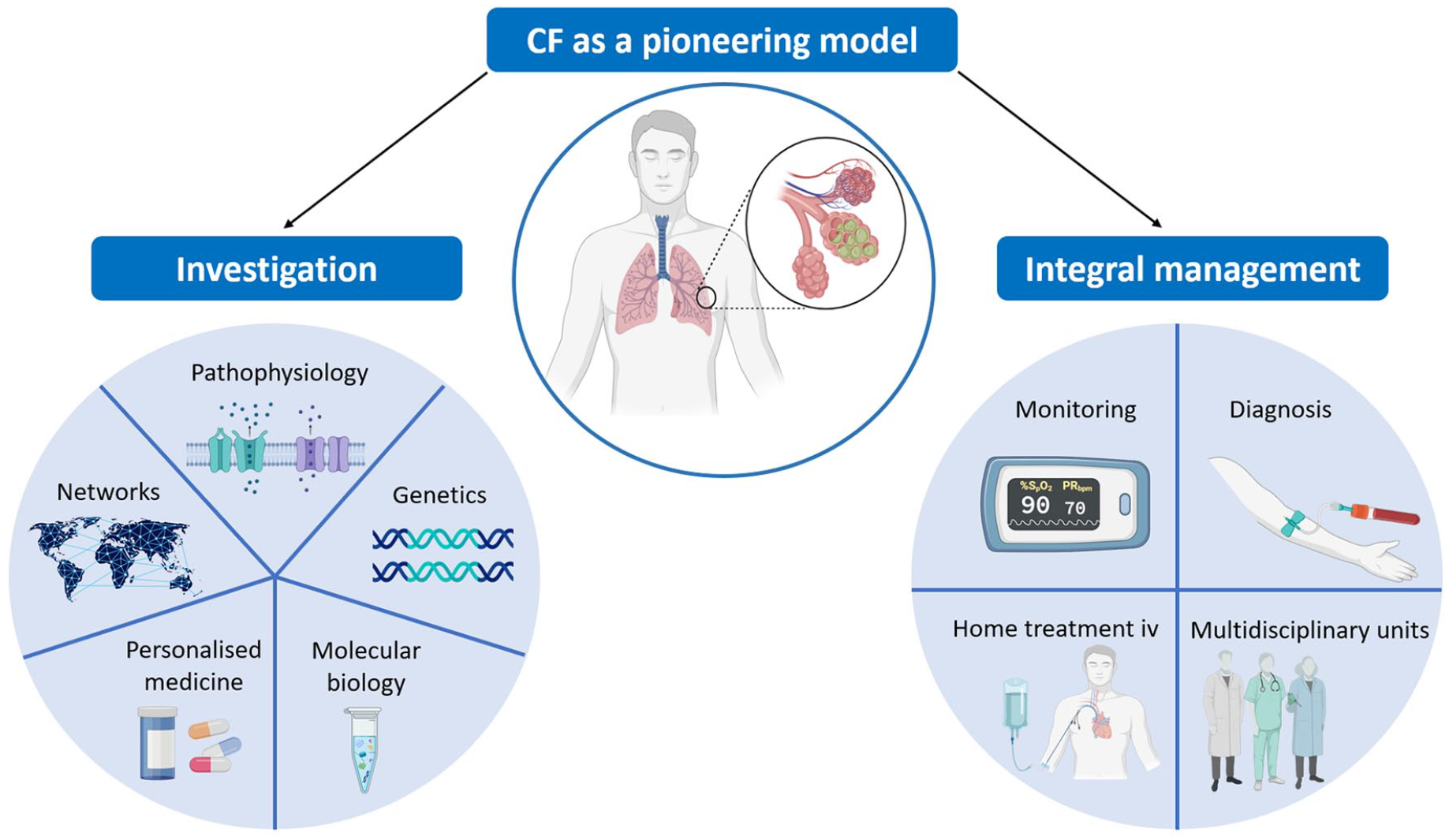
CF a pioneering approach.
First, CF has driven innovation in targeted therapies. The discovery of the CFTR gene in 1989 marked a milestone in genetic medicine, opening the door to the development of specific treatments based on correcting the genetic defect. CFTRm have proven effective in improving lung function and quality of life for pwCF, providing a new paradigm for the treatment of other genetic and respiratory diseases. However, there is still a considerable percentage of pwCF who cannot benefit from this treatment, mainly due to the type of genetic variants. For this reason, other strategies, such as the use of organoids or primary human nasal epithelial cells to test the effectiveness of new treatments, have been explored. 6 The use of organoids in CF, pioneered in the early 2010s, has now extended to many other respiratory diseases, representing a major breakthrough. 7
Lesson 2: Collaborative research networks
The establishment of collaborative research networks and patient registries has significantly enhanced data sharing and the development of multicentre clinical trials, which are essential for accelerating both research and the implementation of new therapies.
A prime example of excellence is the European Cystic Fibrosis Society (ECFS) and the Cystic Fibrosis Foundation (CFF), both recognised for their patient registries and advanced clinical trial networks. The ECFS manages the Clinical Trials Network (ECFS-CTN), established in 2008, which includes 68 clinical research centres, while the CFF has led the Therapeutic Development Network in North America since 1998, with a network of 80 clinical research centres. These entities are known for providing data of the highest quality, well-established standardisation of procedures, specialised training, resource availability, strong collaboration with patient organisations and, finally, due to experienced clinical support of CF teams.
These initiatives have created a replicable model for the study of various respiratory diseases, and their effectiveness in both common and rare conditions. Starting with the integration of genetics, molecular biology and personalised medicine, CF management has laid the foundations for a more holistic approach to the treatment of other respiratory diseases. Examples include the European Reference Network on Rare Respiratory Diseases (ERN-LUNG) for rare respiratory diseases, the International chronic obstructive pulmonary disease (COPD) Coalition for COPD, the International Severe Asthma Registry for Asthma, the Pulmonary Vascular Research Institute for Pulmonary Hypertension and the Global Tuberculosis Network (GTN) for tuberculosis research. These networks facilitate data consolidation and foster collaboration among multiple international centres, accelerating the development of personalised treatments and improving clinical outcomes for these other conditions.
Lesson 3: Pathophysiology of chronic respiratory infections and inflammation
CF research has significantly contributed to a better understanding of the pathophysiology of chronic respiratory infections and inflammation. Since many respiratory diseases share similar pathophysiological mechanisms, it is possible that smoking-related COPD and CFTR function is significantly reduced as a result of prolonged exposure to cigarette smoke. 8 This dysfunction persists even in former smokers, long after they have quit smoking. The alteration in CFTR function critically affects ion transport in the airways, contributing to the perpetuation of inflammation and mucociliary dysfunction. Both factors are key to the progression of the disease, regardless of active exposure to cigarette smoke.
Lesson 4: Expanded clinical trials with medications specifically targeting CFTR
Currently, various clinical trials are being conducted to evaluate CFTR modulators in COPD, such as Icenticaftor, which has shown an increase in FEV1 of between 50 and 60 mL, with a favourable efficacy and safety profile. 9 In patients with bronchiectasis not associated with CF, the efficacy of triple modulator therapy (Elexacaftor/Tezacaftor/Ivacaftor) is also being investigated. 10 These preclinical studies and clinical trials support the pharmacological restoration of acquired CFTR dysfunction as a promising therapeutic strategy that goes beyond CF.
Lesson 5: Management of respiratory exacerbations
The management of respiratory exacerbations in pwCF has been one of the greatest examples of innovation for other respiratory diseases. These exacerbations represent a significant economic burden due to the traditional concept that intravenous treatments require hospitalisation. However, since 1974, when Rucker and Harrison first published their experience with home intravenous antibiotic therapy in children with CF, 11 various intravenous treatment delivery devices, such as elastomeric or electronic infusion pumps and different types of central venous catheters (peripherally inserted central catheter – PICC, midline catheter, subcutaneous port – Port-a-Cath and tunnelled central venous catheter) have been developed, which are now used in other chronic respiratory diseases like COPD or bronchiectasis.
In addition, inhaled antibiotic therapy, mucolytics and associated delivery devices, represent a fundamental therapeutic approach in the treatment of most respiratory diseases associated with chronic bronchial infections (CBI). The direct administration of antibiotics to the airways allows for high local concentrations to be achieved, thereby reducing the risk of systemic effects and enhancing treatment efficacy.
In the context of CF, diluted intravenous formulations were used for nebulised administration under a compassionate use regime for many years, particularly in patients with infections caused by Pseudomonas aeruginosa. Currently, many of these intravenous antibiotics have been formulated for nebulisation, including colistin, tobramycin, aztreonam, levofloxacin and liposomal amikacin. 1
Inhaled antibiotics have also been shown to be an effective and safe treatment option for patients with COPD experiencing multiple exacerbations or with CBI caused by various pathogens, particularly those infected with Pseudomonas aeruginosa. 12
In contrast, the experience with inhaled antibiotics in asthma is limited. This is partly due to airway hyperreactivity in asthma. Therefore, further research is required to determine the safety and efficacy of inhaled antibiotic therapy in these patients.
Moreover, studies on bacterial colonisation and immune responses in pwCF have provided valuable insights into the management of bronchial infections and chronic inflammation in other lung diseases. 13
Lesson 6: CF centre model and multidisciplinary approach
Since the beginning, CF has fostered a comprehensive and multidisciplinary approach to its management, rooted in the CF centre model within the rare disease community. These centres bring together a wide range of professionals into a cohesive team, ensuring essential minimum standards, such as adequate staffing levels and specialised resources. This collaborative model provides personalised and holistic care that addresses not only respiratory symptoms and systemic complications inherent to the disease but also the emerging comorbidities associated with the ageing of this population. This CF centre model has been the axis to improve life expectancy over the last four decades in CF by searching and applying multiple preventive and therapeutic strategies.
However, despite these advancements, there is a concerning trend towards dismantling these centres in favour of more dispersed and fragmented adult care models. This shift could be attributed to insufficient national healthcare resources or a lack of effective communication and coordination between paediatric and adult units. Such fragmented approaches lack the strong identity and collaborative spirit that define CF centres, which have historically been instrumental in facilitating interaction between healthcare professionals, pwCF representatives and hospital administrators. For a truly effective multiprofessional and multidisciplinary approach, it is essential not only to respect and reinforce the diverse roles of medical and allied health professionals but also to put pressure on policy-makers when needed and to acknowledge and value the historical context of the disease and the impact of previous care models.
In COPD the concept of treatable traits has developed considerably during the last decade, aiming to offer a personalised model of care by considering the numerous aspects of the disease in fragile patients. 14 The concept in COPD and other chronic diseases is quite close to the multidisciplinary approach of CF and they could possibly become even more similar in the future of holistic medicine.
The experience gained in coordinating multidisciplinary teams is particularly important in respiratory diseases that affect patients from birth or childhood. Adult care teams must learn from those who provided paediatric care, viewing transition programmes as more than mere bureaucratic processes. Various studies have shown that if this process is not managed properly, it can negatively impact the future health of these young people and increase their morbidity and mortality. The multidisciplinary and transitional approach in CF promotes continuity of care, improves treatment adherence and fosters greater knowledge and independent management of the disease. This model has been successfully implemented in other conditions with complex respiratory involvement, such as severe asthma or neuromuscular diseases but it is likely to be further developed in other diseases such as bronchiectasis – where the gaps between paediatric and adult care settings are still numerous and include the need of a transition protocol. 15
Lesson 7: Innovation in diagnostic and monitoring techniques
CF has been a driving force for innovation in diagnostic and monitoring techniques, such as the use of biomarkers and genetic testing, including newborn screening and advanced genetic sequencing (Next-Generation Sequencing – NGS). Moreover, telemedicine, first applied in CF in 1986, has been pivotal in the development of complementary tools such as portable spirometers, now used both in monitoring this condition and in the follow-up of lung transplants 16 and interstitial lung diseases. 17
More recently, the Multiple Breath Washout (MBW) technique, (first described in 1952 as a method to assess the uniformity of lung ventilation through nitrogen washout,) has gained significant importance in the monitoring of CF. Although it was not initially designed specifically for this condition, its development laid the foundation for its application in subsequent CF studies, especially from the 2000s onwards. Since then, the MBW has become a highly sensitive tool for detecting early stage lung abnormalities, even when spirometry results remain within normal ranges. Currently, the MBW has been progressively incorporated into the assessment of other respiratory conditions, such as primary ciliary dyskinesia (PCD), 18 COPD, 19 asthma 20 and bronchiectasis. 21
Lesson 8: Associations of pwCF
Associations of pwCF have played a crucial role in the evolution of clinical management and research in the disease. 22 These organisations not only provide comprehensive support, educational training and advice to patients and their families but also act as catalysts in promoting research and securing financial resources for the development of specific scientific projects in CF. Moreover, they actively collaborate with specialised CF units, offering logistical and service support according to identified needs and are tireless advocates for patients’ rights before health authorities and government bodies.
The sustained pressure exerted by these associations over the past four decades has been decisive for the rapid and significant progress in the treatment and care of CF. Their influence has been evident in both the shaping of health policies and the direction of scientific research, setting the standard for the creation of regulatory frameworks that support the development of new therapeutic approaches and improving patients’ quality of life. The tireless efforts of these organisations, from the larger associations such as the CFF (USA, established in 1955) and the ECFS (EU, established in 1997), to local initiatives, have served as a model for other groups affected by rare diseases, fostering the organisation of similar movements in defence of their rights and clinical needs.
Conclusion
The management and prognosis of CF have changed radically over time. Lessons learned during this journey can provide a model for the research and management of other respiratory diseases. Adopting similar approaches in other conditions could transform care and significantly improve clinical outcomes across a broad spectrum of chronic respiratory diseases.