
Abstract
Over the past decade, major clinical advances have been made in the healthcare and therapeutic development for cystic fibrosis (CF), a lethal genetic disease caused by mutations in the gene encoding the CF transmembrane conductance regulator (CFTR) protein. CFTR modulators represent innovative treatments that directly target the primary defects in the mutated CFTR protein and have demonstrated significant clinical benefits for many people with CF (pwCF) who are eligible for these treatments. In particular, the triple combination therapy composed of elexacaftor, tezacaftor, and ivacaftor (ETI) has changed the CF therapeutic landscape by significantly improving lung function, quality of life, and predicted survival rates. Here, we provided a comprehensive summary of the impact of ETI on clinical outcomes and the need for further research on long-term efficacy, side effects, pregnancy, possible drug–drug interactions, and extra-pulmonary manifestations. Moreover, a significant number of pwCF are unresponsive to these drugs or cannot afford their high costs. We, therefore, discussed health inequity issues and alternative therapeutic strategies under development aiming to obtain effective therapies for all pwCF.
Cystic fibrosis: Clinical features, diagnosis, and treatment
Cystic fibrosis (CF) is a multisystem disease caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) protein.1,2 It affects more than 100,000 individuals worldwide, with substantial regional variability. It is also considered a lethal genetic disease with high prevalence in people of European descent, although CF also occurs in other populations.3 –5 CFTR variants that cause deficient or dysfunctional CFTR protein disrupt chloride/bicarbonate secretion, sodium reabsorption, and water transport, leading to epithelial surface lining dehydration and mucus hyperconcentration. Typically, CF is characterized by elevated sweat chloride concentration (SCC), obstructive lung disease, chronic bacterial infections of lower airways and sinuses, bronchiectasis, and male infertility due to obstructive azoospermia.1,2
More than 2100 CFTR variants have been recorded (http://www.genet.sickkids.on.ca/app, accessed on January 2, 2025), although to date 1167 are annotated on the CFTR2 website (https://cftr2.org/, accessed on January 2, 2025) and differentiated in CF causing (n. 1085), variants of varying clinical consequence (VVCC) (n. 55), and non-CF-causing (n. 27).
Furthermore, according to their impact on the production, trafficking, functioning, or stability of the CFTR mRNA and/or protein, they are typically categorized into six classes: variants of classes I–III usually result in little to no CFTR activity, leading to potentially severe clinical outcomes and more often pancreatic insufficiency, while variants from classes IV–VI allow significant residual CFTR function leading to milder phenotypes and more often pancreatic sufficiency.6,7 However, a mixture of deficiencies in different CFTR alleles has been reported reflecting the composite defects in mutant CFTR biology.7 –11
The diagnosis of CF is based on positive newborn screening (NBS), clinical features consistent with CF, or a positive family history, in the presence of a high SCC and/or two CFTR-causing variants in trans (i.e., on two different alleles). 12 The age at diagnosis is strongly influenced by the presence or absence of a national or regional CF NBS program. However, over the last decade, the proportion of children diagnosed with CF through NBS has increased to almost 90% throughout Europe, 3 while in the U.S., the median age at first CF clinic visit has decreased to 21 days in 2023. 13 This is a real success given that identifying CF earlier is crucial to prevent the evolution of disease and potential complications. However, a late diagnosis of CF is possible in the presence of recurrent respiratory tract infections and diffuse bronchiectasis, infertility in males, severe sinusitis, or recurrent pancreatitis. These people with CF (pwCF) are more likely to harbor residual function variants or VVCC, in trans with a CF-causing variant, associated with a milder phenotype. 14 Furthermore, a delayed diagnosis with worse clinical outcomes is reported in people of color or living in underprivileged regions. They are more likely to carry (ultra)rare CFTR variants, omitted from CF NBS panels, leading to disparities in care. 15 Until one decade ago, CF management primarily centered on symptom relief and slowing disease progression. The major efforts were on preventing malnutrition and bacterial infections in infants, emphasizing the importance of initiating care as soon as the diagnosis was made. 16 Several chest physiotherapy techniques are known to promote the expectoration of mucus, such as autogenic drainage or using devices that create positive pressure (positive expiratory pressure therapy (PEEP) or high-pressure PEEP therapy) or a vibration (oscillating devices). They are recommended in all pwCF from the first months of life, with positive short-term effects, increasing lung function and sputum expectoration. 16
To date, while the survival and quality of life of pwCF have significantly improved, lung disease remains the most common cause of morbidity and death, especially in pwCF ineligible for CFTR modulators—a novel class of drugs targeting the fundamental defect(s) caused by CFTR pathogenic variants.1,5 Nevertheless, mucoactive drugs remain crucial for reducing secretion build-up, preventing infections, and slowing lung damage. 17 Inhaled antibiotic therapy is still the key for eradicating pathogen bacteria, such as Pseudomonas aeruginosa (Pa), or maintaining long-term control in case of chronic infections. 18
In this paper, a narrative review of the literature was performed, using the MEDLINE/PubMed database, including English-language studies, published from 2019 (i.e., when the first paper on elexacaftor/tezacaftor/ivacaftor (ETI) efficacy was published) to 2024. We aimed to explore the changes in CF with the advent of ETI, the new challenges for clinicians, and future perspectives.
CFTR modulators: Potentiators and correctors approved for clinical use
The introduction of CFTR modulator therapies has revolutionized the course and treatment of CF, impacting the health and quality of life of pwCF. 19 These drugs target defects in the structure and function of the CFTR protein. 5 However, CFTR modulators are only effective for specific variants since they cause different defects in the CFTR protein. On this basis, they have been classified into five main groups: potentiators, correctors, amplifiers, stabilizers, and read-through agents. 5
Until late 2024, there were three correctors and one potentiator available in the market. The potentiator Ivacaftor (IVA; Kalydeco®; VX-770) is effective on variants causing defective gating/conductance by facilitating the opening of the CFTR channel and increasing CFTR-mediated chloride and bicarbonate secretion. 20 In 2022, 1146 (2.1%) out of 54,546 Europeans with CF were eligible for IVA. 3 In the first study on the efficacy of IVA in pwCF carrying at least one G551D variant, it was associated with impressive improvements by 2 weeks and maintained through week 48, with a reduction of SCC of 48.1 mmol/L and positive effects on percent predicted forced expiratory volume (ppFEV1), respiratory exacerbations, weight, and respiratory symptoms. All enrolled adults with CF had an SCC lower than the cutoff point for the diagnosis of CF, which is 60 mmol/L. 21 These results were later confirmed in increasingly younger individuals with other gating variants 22 or with variants with residual function,23,24 so that the indication is currently expanded to include pwCF who are as young as 1 month old and have at least one CFTR variant responsive to IVA based on clinical data, in vitro assay data or both.25,26 Furthermore, in infants aged 4 to <12 months with CF and pancreatic insufficiency, a partial recovery of pancreatic function has been reported. 27
IVA is on the market also in combination with CFTR correctors. These molecules enhance the folding of mutant CFTR protein, rescuing its trafficking to the cell surface.5,28 Lumacaftor (LUM), Tezacaftor (TEZ), and Elexacaftor (ELX) are currently approved in different combinations. The combination LUM-IVA (Orkambi®) has proven moderately effective in F508del-homozygotes individual with an improvement in ppFEV1 ranging from 2.6 to 4.0 percentage points more in the treated group, although some adverse effects have been reported. 29 Furthermore, it led to a weight gain and a significant reduction in the frequency of pulmonary exacerbations (PEx), CF-related hospitalizations, and extra-pulmonary complications such as CF-related diabetes, CF bone disease, and CFTR function, measured as a decline in SCC of 17.8–21.4 mmol/L in real-world settings.30,31 Although these improvements are less relevant than the benefits of IVA in pwCF with gating variants, LUM-IVA was the first hope for F508del-homozygous individuals, who represented 44% of Americans with CF in 2022. 3
Co-treatment TEZ-IVA (Symdeko® or Symkevi®) is effective both in F508del-homozygous individuals and in residual-function heterozygous compounds, thus expanding the possibility of access to CFTR modulators.32,33 Furthermore, TEZ has better pharmacokinetic properties and fewer adverse effects than LUM.32,33 However, it was quickly replaced by the latest and most effective CFTR modulator, that is the triple combination ELX-TEZ-IVA (ETI; Kaftrio® or Trikafta®). In 2019, two randomized, double-blind, placebo-controlled trials reported the impact of ETI on pwCF with at least one F508del, resulting in an improvement of ppFEV1 of 14.3 and 10.0 points after 24 weeks of treatment and a decline of SCC of 41.8 mmol/L and 45.1 mmol/L in F508del/CFTR minimal function and F508del-homozygous individuals, respectively.34,35 Benefits of ETI are also on chest computer tomography (CT) scores, CF-related diseases, inflammatory markers, and circulating levels of fat-soluble vitamins,36 –38 thus significantly transforming the lives of eligible pwCF.39,40
How have CFTR modulators changed the face of CF?
The CF Foundation Patient Registry currently provides updated preliminary data to 2023 on 33,288 pwCF. 13 First of all, the enormous impact of ETI is clear from the increase in the predicted survival 41 and the dramatic decline in the number of pwCF undergoing lung transplants. While the median survival is lower for ineligible pwCF and an individual variability is known, among those born in 2019–2023 it is 61 years, which is almost doubled compared to the years 1999–2003. 13 Furthermore, a recent study by Lopez et al showed a median projected survival for pwCF initiating ETI between the ages of 12 and 17 years to 82.5 years—an increase of 45.4 years compared with best supportive care alone. 41
This is one of the main factors why the number of adults with CF has steadily increased over the past 10 years, representing 60.4% of Americans with CF in 2023. 13 It is, therefore, expected that they will experience increasingly common non-pulmonary complications, which are more prevalent in adults, such as CF liver involvement, CF-related diabetes, osteoporosis, depression, anxiety, cardiovascular diseases, and cancer, among others. 42 Collaboration with specialists from a wide range of disciplines will be necessary, as will greater involvement in primary care.16,43,44
Indeed, before the advent of ETI, CF was one of the major indications for lung transplantation worldwide. Over the last years, most pwCF on ETI with advanced lung disease and listed for lung transplantation were removed from the waiting list. Furthermore, those who were under active evaluation for transplantation listing showed such an improvement that they were no longer considered for it.45,46 Data from the French registry showed that lung transplantation for CF was reduced by 55% in 2020 as compared with 2018–2019. 46 Likewise, in 2023 only 61 lung transplants were reported in the Cystic Fibrosis Foundation (CFF) registry. 13
CFTR modulator therapies may enhance female fertility by improving physical and psychological status as well as by normalizing the cervical micro-environment. 47 For these reasons, the number of pregnancies with CF is continuously increasing. Between 2003 and 2023, pregnancies in women with CF in the U.S. moved from 162 to 675. 13 This highlights the importance of counseling on contraceptive use and family planning before initiation and at routine intervals in ETI therapy.
Registry data also highlight how the prevalence of Pa infections has markedly decreased, from 44% in 2018 to 25% in 2023. 13 Similar data were also for other bacteria typical in pwCF, such as Stenotrophomonas maltophilia (from 12% to 5%), Methicilin-resistant Staphylococcus aureus (from 25% to 14%), Achromobacter xylosoxidans (from 6% to 2%), Burkholderia cepacia complex (from 3% to 1%), and Nontubercolosis mycobacteria (from 14% to 10%).13,48 This trend and the ever-increasing attention to infection prevention and control have also helped reduce the number of PEx in pwCF. 13
The advent of CFTR modulators and, in particular, of ETI has also disrupted some milestones in the clinical course of the disease. Early use of IVA, LUM-IVA, and even more ETI may allow restoration of pancreatic function in pwCF with pancreatic insufficiency, 49 an entity historically considered irreversible. Likewise, pwCF with pancreatic insufficiency on ETI can now develop more often acute pancreatitis due to an increased acinar reserve. 50 Finally, ETI appears to have the potential of reducing the risk of pancreatitis in pwCF with CFTR variants with a residual function by increasing CFTR-mediated chloride secretion. 51
ETI treatment can improve bronchial wall thickening and mucous plugging in studies using magnetic resonance imaging (MRI) 52 and CT, 37 up to complete or partial resolution of cystic bronchiectasis,53,54 always described as permanent dilatation of the airways on high-resolution CT scan.
ETI can also have a beneficial impact on glucose metabolism in individuals with CF-related diabetes, leading to a possible need for reduction or cessation of insulin therapy. 55 These data highlight the need for close surveillance, as well as for the possibility of underestimating the risk of some complications. While CF is commonly associated with malnutrition, 16 in the era of CFTR modulators, overweight and obesity are progressively becoming a significant critical issue, especially in adults with CF. 56 They seem associated with male sex, older age at diagnosis, and pancreatic sufficiency, being also risk factors for cardiovascular and metabolic diseases. 56
Differences in the intestinal microbiome of pwCF compared to healthy controls are known, influencing growth and disease progression in children. Recent reports show that ETI consistently improves microbiome diversity in children, showing taxonomic and functional changes, and reducing measures of intestinal inflammation and antibiotic resistance genes.57,58 Long-term and large case series studies are needed. Multiomic approaches (genomics, metabolomics, microbiomics, and proteomics) have been extensively used to unravel CF pathological mechanisms; however, only a few studies have explored these technologies to identify novel biomarkers associated with CFTR modulator responses (Table 1). Integrative multiomics could add a valuable perspective on how to individualize CF care.
Omics studies assessing the effects of CFTR modulators on samples of people with CF.

ETI, elexacaftor-tezacaftor-ivacaftor; IVA, ivacaftor; LUM-IVA, lumacaftor-ivacaftor.
What are the new challenges for the clinicians beyond elexacaftor/tezacaftor/ivacaftor?
New technologies in healthcare always come with new challenges. CF care has significantly changed, and clinicians must face new dilemmas and question marks (Table 2). Data on the efficacy and safety of ETI, particularly in the pediatric population, remain scarce, highlighting the need for registry or real-world studies reflecting the experiences of pwCF. Clinical trials exclude groups of pwCF with milder or advanced lung disease and include a small number of young children and babies so that regulatory approval for pediatric age is extrapolated on efficacy data obtained in adults with CF. 66 Recent real-world studies show that changes in outcomes can be different from those reported in clinical trials.67–69 Furthermore, among the pediatric clinical trials for ETI, the one by Mall et al. 70 is placebo-controlled and includes the lung clearance index (LCI) as the primary outcome, a much more sensitive tool than ppFEV1 to detect improvements with interventions or significant structural or functional abnormalities. 71 While LCI is a very promising tool for tracking lung function in children or adults with CF with milder disease or normal ppFEV1, it might underestimate lung disease severity in pwCF with established obstructive lung disease.
Priorities and challenges in the era of CFTR modulators.

CF, cystic fibrosis; CFTR, cystic fibrosis transmembrane regulator; ETI, elexacaftor/tezacaftor/ivacaftor; IRT, immunoreactive trypsinogen; LCI, lung clearance index; MRI, magnetic resonance imaging.
A high level of heterogeneity in treatment response to ETI is described. 72 The associated factors are still poorly understood. CFTR modulators require at least 10 g of fat to be taken in order to ensure it is properly absorbed, which is a task not easily achievable in children. Moreover, these molecules are metabolized by cytochrome P450 subtypes CYP3A4 and CYP3A5, which are inhibited by several drugs commonly co-administered in pwCF. 73 New insights also emerge about the influence of variants in cis with F508del in pwCF and their responses to CFTR modulators.74 –76 For instance, the L467F in complex allele with F508del showed no rescue by the LUM-IVA modulator combination 74 and was also unresponsive to ETI therapy74,75 in primary nasal epithelial cells. 76 Clinicians should investigate the presence of cis variants in pwCF with reduced or absent response to modulators, as well as ensure correct absorption of the drug or incompletely understood drug–drug interactions that may interfere with outcomes. It would be even more desirable to predict the CFTR modulator’s efficacy in patient-derived tissues, such as primary bronchial cells, primary nasal cells, or rectal biopsy-derived organoids. First data on the use of plasma quantitation of ETI have been reported, 77 aiming to optimize the therapeutic effect and the drug exposure, minimizing adverse events.
The majority of pwCF in clinical trials experienced adverse events.34,35 Most of them were mild or moderate in severity, consistent with CF manifestations or common childhood infections. However, close monitoring is necessary especially for liver tests, given that an increase in transaminases is observed in approximately 10%–26% of children69,70 and 5% of adults 78 on ETI therapy, causing a suspension of the drug very rarely. 69 In pwCF with liver involvement, ETI is associated with improvement in liver stiffness and fibrosis indices, probably reducing the development of advanced CF liver disease. 79 Longer observations with larger cohorts are needed to confirm this data.
Furthermore, ETI may significantly increase serum cholesterol by enhancing its absorption, as evidenced by a correlation between serum cholesterol and phytosterol levels.80,81 These metabolic changes and the increased prevalence of overweight and obesity among pwCF (from 10% to 21% among children with CF and from 18% to 42% among adults) 13 may lead to a higher risk of co-morbidities, such as metabolic disorders and cardiovascular disease, especially with chronic administration in the pediatric population.
Worrying albeit rare adverse events have also been reported, such as raised intracranial pressure,69,82 neuropsychiatric effects on mental health,83,84 drug reaction with eosinophilia and systemic symptoms like syndrome, musculoskeletal injuries, or severe decrease in platelet count. 85 Research is also continuing in pwCF eligible for ETI, as demonstrated by recent data on vanzacaftor–tezacaftor–deutivacaftor (VTD), a once-daily dosing combination with fewer drug interactions than ETI. 86 While recent data from phase III trials ensured the approval of VTD as a novel therapy for pwCF harboring at least one F508del allele or another responsive CFTR variants,87,88 the long-term effects remain to be investigated.
Even with the recently published evidence-based standards of care for CFTR variant-specific therapies, 89 understanding biological mechanisms underlying adverse events, optimizing the drug exposure or early identification of pwCF at greatest risk of adverse events will be among the major challenges of coming years.
Little data are yet available on long-term efficacy. Daines et al. demonstrated in children aged >12 years and adults with CF in ETI at week 144, durable clinical benefits, with a good safety profile. 90 Nevertheless, in a 5-year single-center retrospective study, pwCF who received IVA showed an acute improvement in lung function, with a subsequent decline and the need to optimize care again. 91 Clinicians must closely monitor the lung function, SCC, and nutritional status of pwCF in ETI, especially for those with advanced lung disease (ppFEV1 ⩽ 40% or waiting for a lung transplant) before starting the drug, and efforts must be directed to maintain high adherence with treatments. In fact, concerning overall improvement after CFTR modulators, the risk of reduced treatment adherence is a critical factor.92,93 Reducing the therapeutic burden is a priority for pwCF, especially for those with mild-moderate lung disease. 39
The SIMPLIFY study demonstrated that in pwCF undergoing ETI and having a relatively well-preserved pulmonary function, discontinuing daily hypertonic saline or dornase alfa for 6 weeks did not lead to clinically significant differences in pulmonary function compared to continuing treatment.94,95 However, the follow-up period was very short and included pwCF had very mild lung disease, with a ppFEV1 of 96.9%. Other ongoing projects, such as CF STORM (52-week trial) and NEEMO (prospective observational study with 12-month assessments up to 5 years) will address the safety of stopping additional treatments in pwCF on ETI.
Nevertheless, while IVA treatment resulted in a decrease in sputum Pa density and total bacteria burden, the infection rebounded after 1 year of the drug and with the same strain of bacterium. 96 Likewise in pwCF in ETI, Pa remains detectable by amplicon sequencing throughout the respiratory tract.97,98 For this reason, a recent paper by experts on this focus concluded that pwCF on CFTR modulators may still need inhaled antibiotics to maintain long-term control of Pa infections. 19 Furthermore, another challenge in the era of CFTR modulators, especially with ETI, is the definition of Pa chronic infection. Many pwCF on ETI fail to produce sputum, being needed to monitor airway infections. Such investigations include the use of induced sputum, for pwCF colonized by Pa before starting the drug and with multiple negative cultures indicating a possible clearance. A clear guideline is desirable on this topic.19,99
Caution and careful monitoring are necessary in case of infants exposed to ETI in utero and while breastfeeding. 47 More cases are reported on the safety of ETI in pregnancy 47 ; for instance, congenital bilateral cataracts in newborns exposed to the drug have been reported, highlighting the need for cataract screen at birth. 100 Furthermore, decreased blood immunoreactive trypsinogen (IRT) levels in newborn carriers exposed to ETI have been shown, 101 as a case of false negative CF NBS in a CF child born from a mother taking CFTR modulator therapy during pregnancy. 102 Passing ETI through the placenta and into breast milk can have beneficial effects by preventing pancreatic insufficiency and even resolving meconium ileus in fetuses with CF. 103 This implies caution in the interpretation of IRT values and SCC in infants exposed to the drug.
Finally, the increased projected survival for pwCF starting ETI at a young age has brought attention to the risk of colorectal cancer. 104 Lack of CFTR expression promotes carcinogenic processes such as intestinal inflammation and deleterious gut microbiome changes. For this reason, the CFF Foundation recently recommended treating this population as a high-risk group, using a colonoscopy-only screening strategy starting at age 40 in pwCF without transplant and at age 30 after transplant. Screening should be considered every 5 years if negative and every 3 years or sooner for individuals with adenomatous polyps. 104
Health inequities to access to CFTR modulators
While ETI has become a standard of care for children aged 2 or 6 years and older in most high-income countries, 86 significant global disparities persist in access to these life-saving drugs, especially due to the high cost of the drug.105 –108 It is clear that the age of starting ETI impacts clinical outcomes and survival.41,66 A study in Canada showed the projected benefits of ETI on the number of deaths would be halved if the access were delayed to 2025, compared to 2021. 109
The different access possibilities for pwCF and the reimbursement of these drugs are regulated by national regulatory agencies, such as the European medicines agency (EMA) in Europe and by the FDA in the U.S., based on results from phase III double-blind Randomized controlled trials (RCTs). However, there are differences in the extension indications (Table 3). For instance, the FDA has accepted pre-clinical evidence, considering a recovery of the CFTR function ⩾10% based on chloride transport; on the contrary, EMA does not deem preclinical data sufficient to expand the label of CFTR modulators without confirmatory clinical data.107,110 So initial approvals and regulatory review process are expected to be faster in the U.S., where CFTR modulators are approved 267 days earlier than in the European Union. 111 So in most European countries, ETI is prescribed only in pwCF aged ⩾2 or 6 years, with at least one F508del, while in the U.S., pwCF with 271 other responsive CFTR variants are eligible. However, in March 2025 an extension was approved, making ETI prescribable in Europe in all pwCF aged 2 years and over, with at least one non-class I mutation (Table 3). Cromwell et al have recently reported meaningful improvements in lung function in Americans with CF on ETI, carrying 92 out of these 177 CFTR variants, corresponding to 5.2% of U.S. Registry participants. 112 Similarly, the French compassionate program showed that ETI is effective in a large number of pwCF with rare CFTR variants, including variants that were not previously approved by the FDA.113,114
CFTR modulators in people with CF (updated to March 2025).

CFTR, cystic fibrosis transmembrane regulator; EMA: European Medicines Agency; FDA, food and drug administration.
Even more worrying, although the situation moves rapidly, ETI is not yet licensed and reimbursed in several low-middle income European countries or Central and South America, India, the Middle East, and Southern Africa, while in other countries pwCF have access via a mechanism called “a named patient supply program” (Table 4). In other countries, “legal wars” are necessary to obtain the drug, with delays in starting therapy, also due to the difficulty of carrying out the CFTR genetic test. This may force families to migrate to countries where legislative guidelines allow economic coverage of the drug. Race, ethnic diversity, or the knowledge of regional epidemiology and information on CFTR eligible variants are crucial factors influencing eligibility for CFTR modulators, penalizing low- and middle-income countries or favoring populations where F508del is common, such as the U.S. and Northern European populations. 105 In 2023, only 8% of the American population was ineligible for a modulator based on their genotype. 13 However, in 2022, Guo et al estimated that only 12% of the world’s total CF population is receiving these drugs. 4 In countries with mixed ethnicity or with a low prevalence of F508del, such as Brazil 115 and Italy, 16 where over 50% and 30% of pwCF, respectively, have a CFTR variant other than F508del, a larger number of pwCF may not be eligible for CFTR modulators. They have significant physical, mental, and social impacts from their disease and, as recently reported, the current therapeutic options have left them feeling scared, forgotten, and neglected. 116 Clinicians must consider these aspects, and increasingly greater psychological support will be desirable, especially in adolescence. These inequities are added to the already known worldwide differences about the prescription ETI post liver transplantation 117 or to the absence or limited neonatal screening programs in some areas 118 or to the different access to liver and lung transplants. 43 For these reasons, in 2024, differences in patient clinical status and survival could be attributable not only to intrinsic disease severity but also to these disparities. The delay and unlikely universal availability of CFTR modulators in the next years will lead to further widening of global disparities in CF outcomes. 103
Countries where elexacaftor/tezacaftor/ivacaftor (ETI) was licensed and reimbursed as of the 13th December 2024 and age for ETI access (https://www.vrtx.com/medicines/cystic-fibrosis-facts-and-figures/).
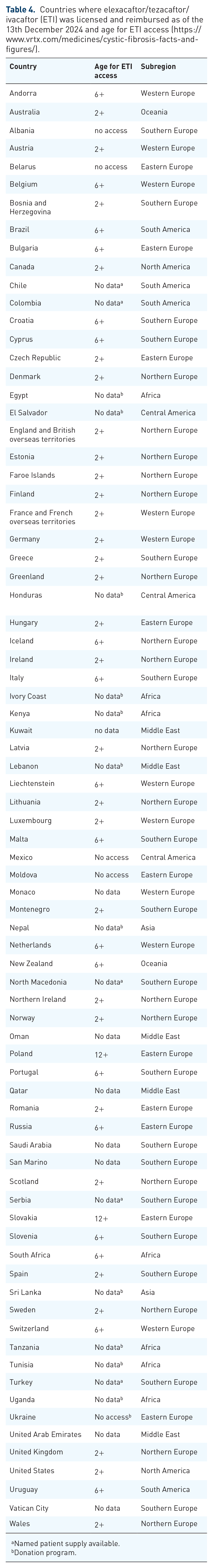
Named patient supply available.
Donation program.
Accordingly, there is an urgent need to reduce the cost of CFTR modulators to negotiate agreements also in low- and middle-income countries. As reported by Guo et al., the annual estimated minimum production cost of ETI is 5676 $–90% lower than the current market price (>300,000$ in the U.S.). 119
What are the future perspectives beyond elexacaftor/tezacaftor/ivacaftor?
Despite the therapeutic success of currently approved CFTR modulators, particularly IVA and ETI, the complete restoration of the function in the lungs and other affected organs is not achieved by these drugs. Indeed, experimental evidence revealed that these drugs are unable to fully recover CFTR protein stability and traffic defects as well as channel gating activity for several variants, including F508del and G551D.20,120 –123 In intestinal organoids and primary nasal epithelial cells, improvement of CFTR function attains only ~50% with ETI treatment, indicating that there is still scope for further improvement. 124 Accordingly, pwCF still face a progressive deterioration of their lung function and complications, albeit at a slower pace. 125 Moreover, several CFTR variants are only moderately rescued (below clinically relevant levels) or unresponsive at all for the currently available modulator therapies. Individuals carrying these CF genotypes will be “bad” or only “mild” responders in whom disease progress will remain at an accelerated pace. 126
While the identification of specific therapies for (ultra)rare CFTR variants poses substantial challenges due to the high variability of CFTR variants and the small number of individuals carrying these genotypes, the implementation of “theratyping” (i.e., matching single variants with effective modulators) has significantly facilitated the characterization of CFTR variants in which the molecular defect(s) was(were) understudied as well as the assessment of available CFTR modulator efficacy to expand their benefit for pwCF carrying (ultra)rare genotypes.127,128 Translational models using patient’s own cells have been developed and these consist of useful tools to understand and predict clinical responsiveness at an individual level. Among those, the most suitable models are represented by the 2D and 3D cultures of nasal epithelial cells and rectal biopsies-derived intestinal organoids. 129 In addition to recapitulating several features of the parental organ, these models were demonstrated to be practical, reliable, and easy to obtain and handle. 129 Indeed, various studies have reported good correlations of the data on patient-derived cell models with clinical features of pwCF (before and after starting ETI therapy).130 –138
In parallel, the CF drug discovery pipeline continues to expand with various small molecules under investigation in early-stage clinical studies. For instance, Sionna Therapeutics is initiating two phase I trials of two CFTR correctors (SION-719 and SION-451), which are described as highly potent NBD1 stabilizers. The company has also signed an agreement with AbbVie to expand and accelerate the development of the corrector ABBV-2222 and the potentiator ABBV-3067 that present complementary mechanisms to Sionna’s compounds. Fair Therapeutics will assess the safety and efficacy of the triple combination composed by the amplifier PTI-428, the corrector PTI-801, and the potentiator PTI-808 in pwCF carrying rare genotypes. The amplifier PTI-428 was found to selectively enhance CFTR translation by stabilizing CFTR mRNA, 139 while the corrector PTI-801 was demonstrated to share a common mechanism to ELX to rescue F508del-CFTR. 123 The individuals enrolled in this clinical study were selected based on their intestinal organoid responses to these CFTR modulators. Novartis has developed the CFTR potentiator icenticaftor, which demonstrated clinical benefit for patients with chronic obstructive pulmonary disease in a phase II study. 140
Several other CFTR correctors and potentiators are in pre-clinical studies with promising results. The corrector ARN23765 demonstrated picomolar potency and lengthened CFTR rescue in vitro at concentrations 5000 lower than LUM. 141 Rescue of F508del-CFTR processing and function was enhanced by the combination of TEZ with the corrector MCG1516A. 142 The compound 4172 and its analogs, in combination with LUM and the corrector 3151, increased the processing and function of several CFTR variants. 143 IDOR-4 is a macrocyclic CFTR corrector that demonstrated a complementary mechanism of action to clinically approved CFTR modulators. 144 Apigenin, Asp-11, CP-628006, and LSO-24 are CFTR potentiators from different chemotypes that were proposed to potentiate CFTR gating activity by a different mechanism than that of IVA.143 –148
Another class of small molecules under development for CF are the read-through agents. These compounds enable the translation machinery to “over-read” a premature termination codon (PTC), thus allowing the insertion of an amino acid to continue the translation to the normal end of the transcript. 20 ELX-02 demonstrated promising read-through activity in vitro,149,150 but clinical data of pwCF carrying G542X taking this compound exhibited only modest improvement in lung function. TLN468 is another agent that allowed the expression of several nonsense CFTR variants in which processing and function were enhanced by the combination with CFTR modulators. 151 SRI-41315 is suggested to induce read-through activity by pausing protein translation at PTCs due to eRF1 suppression. 152 Another alternative for these variants consists in the design of synthetic tRNAs able to decode PTCs, thus introducing an amino acid into the nascent polypeptide chain that enables the translation of the full-length protein. This approach can rescue both mRNA and full-length protein expression.153 –155 Finally, antisense oligonucleotides (ASOs) represent an additional option under development not only for nonsense variants but also for those with splicing defects. 156 These chemically modified synthetic RNA-like molecules act by complementary base pairing to the target sequence and demonstrated promising results for CFTR variants, including W1282X and 3849 + 10 kB C>T.156 –159 As ASOs are very small and considerably stable, it is expected that they could be delivered without the need for a vehicle.
A growing number of studies have demonstrated the feasibility of gene therapy using CFTR cDNA or mRNA to enable patient cells to produce a functional copy of CFTR.160 –165 The major advantage of these approaches is the possibility of using them in a variant-agnostic manner (i.e., anyone with CF could receive this treatment regardless of the underlying CFTR variants). Some promising gene therapeutic products include the 4D-710 (from 4D Molecular Therapeutics), SP-101 (Spirovant Sciences), ARCT-032 (from Arcturus Therapeutics), RCT2100 (from ReCode Therapeutics) and VX-522 (from Vertex Pharmaceutics). The 4D-710 and SP-101 are adeno-associated viral vectors containing WT-CFTRΔR cDNA. The SP-101 demonstrated to restore CFTR-mediated chloride transport in primary human airway epithelial from pwCF, 166 while the 4D-710 is being assessed in adults with CF ineligible or unable to tolerate adverse events of currently available modulator therapies. On the other hand, ARCT-032, RCT2100, and VX-522 are lipid nanoparticles containing the CFTR mRNA that can be delivered to the airway by aerosolization.
Other strategies to directly edit and repair the mutated DNA include homology-directed repair165,167 –169 and base or prime editing using clustered regularly interspaced short palindromic repeated and associated proteins (CRISPR/Cas).170 –174 The ultimate goal of these approaches is to correct cells in which CFTR is expressed and has a fundamental role in the epithelial surface lining homeostasis or to reprogram stem cells that could be administered in vivo to differentiate and repopulate the epithelium. 20 While gene editing holds substantial promise with several publications demonstrating its feasibility, there are still various obstacles to translating this approach into the clinical scenario for CF.
Finally, modulation of alternative (non-CFTR) channels/transporters has been investigated as a potential approach to circumvent the absence of CFTR-mediated chloride and bicarbonate secretion. Some of the main targets include ENaC, TMEM16A, SLC26A4, SLC26A9, and ATP12A.20,175 By inhibiting or enhancing the functional expression of these channels/transporters, it is expected that the epithelial surface liquid homeostasis could be restored. This approach can also serve as CFTR variant-agnostic therapeutics.
Conclusion
It is undeniable that “highly effective” modulator therapies have revolutionized the lives of many pwCF by transforming what was described as an early-age lethal disease into a more manageable chronic condition. However, these therapeutic regimens have brought new dilemmas and questions since these individuals are presenting a healthier profile, and more accurate and sensitive biomarkers will be needed to stratify subtle lung disease alterations and monitor disease progression. Several approaches are under investigation with LCI, CT, and MRI of the lungs already demonstrating to be sensitive measurements for young children and adults with mild disease. Moreover, as pwCF are also living longer, there is an increasingly appreciated need for further studies of the appearance of extra-pulmonary manifestations (CF-related diabetes, osteoporosis, intestinal cancer, and others) and the impact of CFTR modulators on these.
Theratyping efforts remain vital to expand the access of currently available modulator therapies to a greater CF population since a significant number of pwCF carry (ultra)rare CFTR variants and these are usually excluded from traditional clinical trial designs. However, there are also pwCF-carrying CFTR variants that are refractory for these therapies. These include not only canonical splice, PTC, frameshift, and large deletion/insertion variants, but also missense variants with folding and/or gating defects that are unresponsive to currently available modulators. Therefore, alternative therapeutic strategies are required to overcome such defects. While there are several obstacles to overcome in these alternative strategies, significant progress has also been made to translate them into clinical practice. The joint effort of the entire CF scientific community is the most complete way to have effective therapies for all pwCF.