
Abstract
Background:
The antifibrotic drugs nintedanib and pirfenidone are used for the treatment of idiopathic pulmonary fibrosis (IPF). We analysed the association of common profibrotic polymorphisms in MUC5B (mucin 5B, rs35705950) and DSP (desmoplakin, rs2076295) on antifibrotic treatment outcomes in IPF.
Methods:
MUC5B rs35705950 and DSP rs2076295 were assessed in IPF patients (n = 210, 139 men/71 women) from the Czech EMPIRE registry and age- or sex-matched healthy individuals (n = 205, 125 men/80 women). Genetic data were collated with overall survival (OS), acute exacerbation episodes, worsening lung function and antifibrotic treatment.
Results:
We confirmed overexpression of the MUC5B rs35705950*T allele (55.2% versus 20.9%, p < 0.001) and the DSP rs2076295*G allele (80.4% versus 68.3%, p < 0.001) in IPF compared with controls. On antifibrotic drugs, lower mortability was observed in IPF patients with DSP G* allele (p = 0.016) and MUC5B T* allele (p = 0.079). Carriers of the DSP rs2076295*G allele benefitted from nintedanib treatment compared with TT genotype by a longer OS [hazard ratio (HR) = 7.99; 95% confidence interval (CI) = 1.56–40.90; p = 0.013] and a slower decline in lung function (HR = 8.51; 95% CI = 1.68–43.14; p = 0.010). Patients with a TT genotype (rs2076295) benefitted from treatment with pirfenidone by prolonged OS (p = 0.040; HR = 0.35; 95% CI = 0.13–0.95) compared with nintedanib treatment. Both associations were confirmed by cross-validation analysis. After stratifying by MUC5B rs35705950*T allele carriage, no difference in treatment outcome was observed for nintedanib or pirfenidone (p = 0.784). In the multivariate model, smoking, age, forced vital capacity (FVC) and DLCO (diffuse lung capacity) at the IPF diagnosis were associated with survival.
Conclusion:
Our real-world study showed that IPF patients with MUC5B T* allele or DSP G* allele profit from antifibrotic treatment by lower mortability. Moreover, carriers of the DSP rs2076295*G allele benefit from treatment with nintedanib, and TT genotype from treatment with pirfenidone. MUC5B rs35705950 did not impact the outcome of treatment with either nintedanib or pirfenidone. Our single-registry pilot study should be confirmed with an independent patient cohort.
Introduction
Idiopathic pulmonary fibrosis (IPF) is characterised by progressive fibroproliferative healing, occurring mainly in genetically predisposed older individuals. 1 More men than women have been reported with IPF, and most patients have a history of cigarette smoking. 1 To date, several genetic variants in genes coding surfactant proteins, mucin, cytokines, telomerase and senescence-related molecules have been linked to IPF.2,3
Several described gene variants have been shown to influence clinical outcomes and therapeutic responses in IPF.2,4 A recent study showed that carriers of a TT genotype for TOLLIP (rs3750920), but not MUC5B genotypes, benefitted from treatment with oral N-acetylcysteine, resulting in a decreased risk for the composite endpoints of death, hospitalisation or a 10% decrement in forced vital capacity (FVC). 5 Patients with a TERT genotype associated with short telomere length may benefit from telomere-directed therapy. 6 It is not yet clear whether common profibrotic variants in MUC5B (mucin 5B) and DSP (desmoplakin) may affect the outcome of antifibrotic therapy in IPF. A promoter variant in MUC5B rs35705950 is the most substantial genetic risk factor for the development of IPF.7,8 The MUC5B rs35705950 minor T allele is associated with increased activity of MUC5B promoter contributing to the enhanced expression of mucin 5 in the IPF lung, particularly in the bronchioloalveolar epithelia. 9 Also, DSP rs2076295 variants contribute to IPF pathogenesis through the enhanced expression of desmoplakin, 10 a protein vital for structural integrity at adjacent cell contacts, cell–cell adhesion, wound repair and epithelial barrier function.7,11 DSP rs2076295 *G alleles are associated with decreased gene expression in IPF lung tissue compared with the TT genotype. 10
Therefore, we investigated the association of two common gene polymorphisms associated with IPF, the MUC5B and DSP genes, with treatment outcomes in a real-world cohort of patients from the Czech section of the EMPIRE (the European MultiPartner IPF Registry) who had been treated with nintedanib or pirfenidone. The evaluation of treatment outcome, as assessed by overall survival (OS) and decline in lung function, was performed on subgroups treated only with these two approved antifibrotic drugs. Identification of genetic variants associated with different treatment outcomes can help in selecting patients for specific antifibrotic drug therapies.
Materials and methods
Participants
A total of 210 consecutive patients with IPF (139 men/71 women; median age at diagnosis = 70 years, minimum–maximum = 52–82 years) from the Czech section of the EMPIRE registry donated peripheral blood samples for genetic testing. All patients met the American Thoracic Society (ATS)/European Respiratory Society (ERS) IPF criteria.1,12 A total of 167 (79.5%) patients were receiving antifibrotic drugs and 43 (20.5%) received other treatment, as they did not fulfil the local criteria for antifibrotic treatment. 13 Of patients on antifibrotic drugs, 127 (76.0%) were taking pirfenidone and 40 (24.0%) nintedanib. For detailed characteristics of the patients, see Tables 1 and 2. The median follow-up of enrolled patients was 34.3 months; the end-point event was the date of the last clinical examination or death. Reasons for death are shown in Table 3, and the leading cause of death was the progression of IPF (respiratory failure). Acute exacerbations of IPF during follow-up time was observed in 49 IPF patients (23.3%) (Table 2). In addition, 205 age- and sex-matched individuals (125 men/80 women; median age = 70 years, range = 51–88 years) were recruited from care home residents, where chronic lung disease, autoimmunity and cancer were ruled out using patient records.
Distribution of genotypes, alleles and phenotypes (allele carriers, carriage) of investigated MUC5B and DSP variants in the group of IPF patients and healthy control participants.
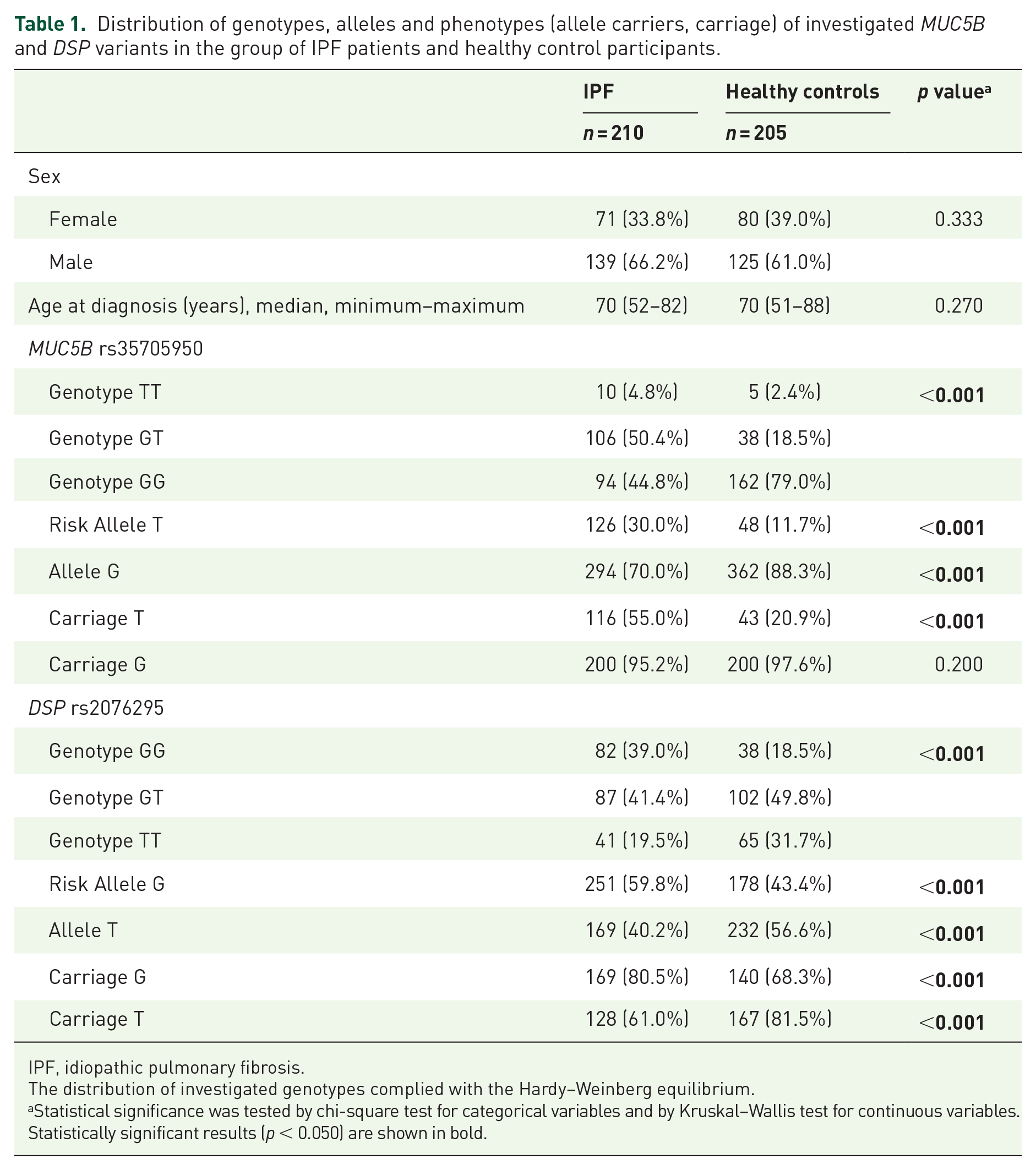
IPF, idiopathic pulmonary fibrosis.
The distribution of investigated genotypes complied with the Hardy–Weinberg equilibrium.
Statistical significance was tested by chi-square test for categorical variables and by Kruskal–Wallis test for continuous variables.
Statistically significant results (p < 0.050) are shown in bold.
Patient characteristics in IPF patients subdivided according to MUC5B rs35705950 and DSP rs2076295 genotypes.

Diff. diag., the difference between the FVC or DLCO at diagnosis and given timepoint; DLCO, diffuse lung capacity; FVC, forced vital capacity; HRCT, high-resolution computed tomography; IPF, idiopathic pulmonary fibrosis; UIP, usual interstitial pneumonia.
Statistical significance was tested by chi-square test for categorical variables and by Kruskal–Wallis test for continuous variables.
Reason of death.

IPF, idiopathic pulmonary fibrosis.
Written informed consent was obtained from all enrolled participants, and the study was in accordance with the Helsinki Declaration and approved by the Institutional Review Board of Thomayer Hospital, Prague.
Monitored parameters
For all IPF patients, demographics (age and sex), clinical characteristics (onset and severity of symptoms, smoking status), histopathology, pulmonary function (forced expiratory volume in 1 s – FEV1, FVC and lung diffusion capacity for carbon monoxide – DLCO) and radiology (high-resolution computed tomography – HRCT) data and bronchoalveolar lavage (BALT) parameters were collected.
Histopathological findings
In all patients who underwent lung biopsy, histopathological pattern according to the ATS/ERS statement has been determined. 1
Pulmonary function tests
Spirometry, body plethysmography and lung diffusion capacity were measured according to the ATS/ERS recommendations. 1 Results were shown as a percentage of predicted values (% pred). Changes in FVC greater than 10%, and in DLCO greater than 15%, were considered to be clinically significant. 14
Chest HRCT
All patients underwent HRCT scanning at diagnosis. Gay’s scoring system (modified by Dutka and Vasakova) was used.15,16 HRCT alveolar and interstitial changes (alveolar = 0–5; interstitial = 0–5) were evaluated in all patients (higher score values correspond to greater extent of the lesion) by a pneumologist experienced in radiology.
Bronchoalveolar lavage
Bronchoalveolar lavage fluid (BALT) was analysed at the time of diagnosis. Physiological findings were defined as follows: lymphocytes < 15%, neutrophil granulocytes < 3%, eosinophil granulocytes < 1% and macrophages > 85%. 17
Genetic analysis
MUC5B SNP rs35705950 and DSP SNP rs2076295 were analysed by targeted next-generation sequencing (NGS) on genomic DNA in both patient and control groups, as reported previously.18,19 Amplicon-based libraries were sequenced as paired end on MiSeq (2 × 151 bp, Illumina, San Diego, CA, USA).
Statistics
Descriptive statistics were used to summarise the demographics and clinical characteristics of the IPF patients at enrolment. 20 For the analysis of survival, the Kaplan–Meier method was used to estimate the cumulative incidence function of time from diagnosis until death from any cause. The Cox proportional hazards regression model was used to identify alleles as prognostic factors of survival. The models were validated by k-fold cross-validation. Data were analysed using IBM SPSS 25 for Windows (Release 25, IBM Corporation 2013), Stata/IC 14.2 for Windows (Stata Statistical Software: Release 14. College Station, TX: StataCorp LP) and R (version 3.6.3).
Results
MUC5B rs35705950 genotypes and DSP rs2076295 genotypes in IPF patients
Distribution of the MUC5B rs35705950 (IPF risk allele T) and DSP rs2076295 (IPF risk allele G) genotypes and allele frequencies in IPF patients and control participants are shown in Table 1. Distribution of the investigated alleles complied with the Hardy–Weinberg equilibrium.
Regarding MUC5B rs35705950, a GT genotype (50.4% versus 18.5%, p < 0.001) and carriage T* allele (55% versus 20.9%, p < 0.001) were more common in IPF patients compared with the control group. There was no significant difference in the frequency of rs35705950 GG and TT genotypes between these groups. The MUC5B rs35705950 *T allele was more common in men than women (75.0% versus 25.0%; p = 0.003). In a whole cohort, MUC5B rs35705950 *T allele was associated with worse DLCO at month 18 (p = 0.005) and progression as assessed by DLCO decline of 15% or more (p = 0.048 at month 12, p = 0.061 at month 18). The association of MUC5B rs35705950 *T allele with better FVC (p = 0.085 at month 18) and slower progression as assessed by FVC decline of 10% or less (p = 0.096 at month 18) did not reach significance. BALT neutrophils over 3% were found in 88.9% of TT genotype patients compared with lower numbers in those with GG and GT genotypes (p = 0.025). There was no difference in acute exacerbation episodes, radiological findings and other studied parameters between MUC5B rs35705950 *T allele carriers and patients with GG genotype (Table 2).
Regarding DSP rs2076295, a GG genotype (39.0% versus 18.5%, p < 0.001) and G* allele carriage (80.5% versus 68.3%, p < 0.001) were overrepresented in IPF patients compared with the control healthy group. A DSP rs2076295 TT genotype was less frequent in the IPF group than in controls (19.5% versus 31.7%; p < 0.001). In a whole patient cohort, the presence of DSP G* allele was associated with a radiological diagnosis (p = 0.002), and there was a trend of association with an interstitial score (p = 0.064), but not with sex and pulmonary function parameters (Table 2).
Influence of MUC5B variants on prognosis of IPF and antifibrotic treatment outcome
After stratifying by rs35705950 MUC5B, here was a trend of lower mortality in IPF patients with MUC5B rs35705950 T* allele treated with antifibrotic agents (pirfenidone or nintedanib) compared with those not receiving antifibrotic treatment (p = 0.079) (Figure 1). There was no difference in mortality between IPF patients with the GG genotype treated with/without antifibrotic drugs (p = 0.678). When evaluating both antifibrotic drugs separately, patients carrying the *T allele did not differ in survival when treated with nintedanib [p = 0.795; hazard ratio (HR) = 0.81; 95% confidence interval (CI) = 0.16–4.12) or pirfenidone (p = 0.240; HR = 1.43; 95% CI = 0.79–2.61) compared with those with a GG genotype, as demonstrated by the Kaplan–Meier curve (Figures 2 and 3). No difference in OS was observed between patients carrying the *T allele (p = 0.785; HR = 0.88; 95% CI = 0.35–2.22) or having a GG genotype (p = 0.528; HR = 0.64; 95% CI = 0.15–2.0) when treated with nintedanib or pirfenidone (Figure 1, Table 2). Similarly, no difference in lung function decline was observed between IPF patients treated with nintedanib or pirfenidone in subgroups that were subdivided based on MUC5B variants (Figure 2).

Mortality in IPF patients subdivided according to the carriage of MUC5B*T allele treated with (a) nintedanib or pirfenidone or (b) without antifibrotic treatment and comparison between nintedanib or pirfenidone treatments versus without antifibrotic treatment for (c) *T allele carriers and (d) GG genotypes.

Mortality in IPF patients subdivided according to the carriage of MUC5B*T allele treated with (a) nintedanib or (b) pirfenidone and comparison between nintedanib and pirfenidone treatments for (c) *T allele carriers and (d) GG genotypes.
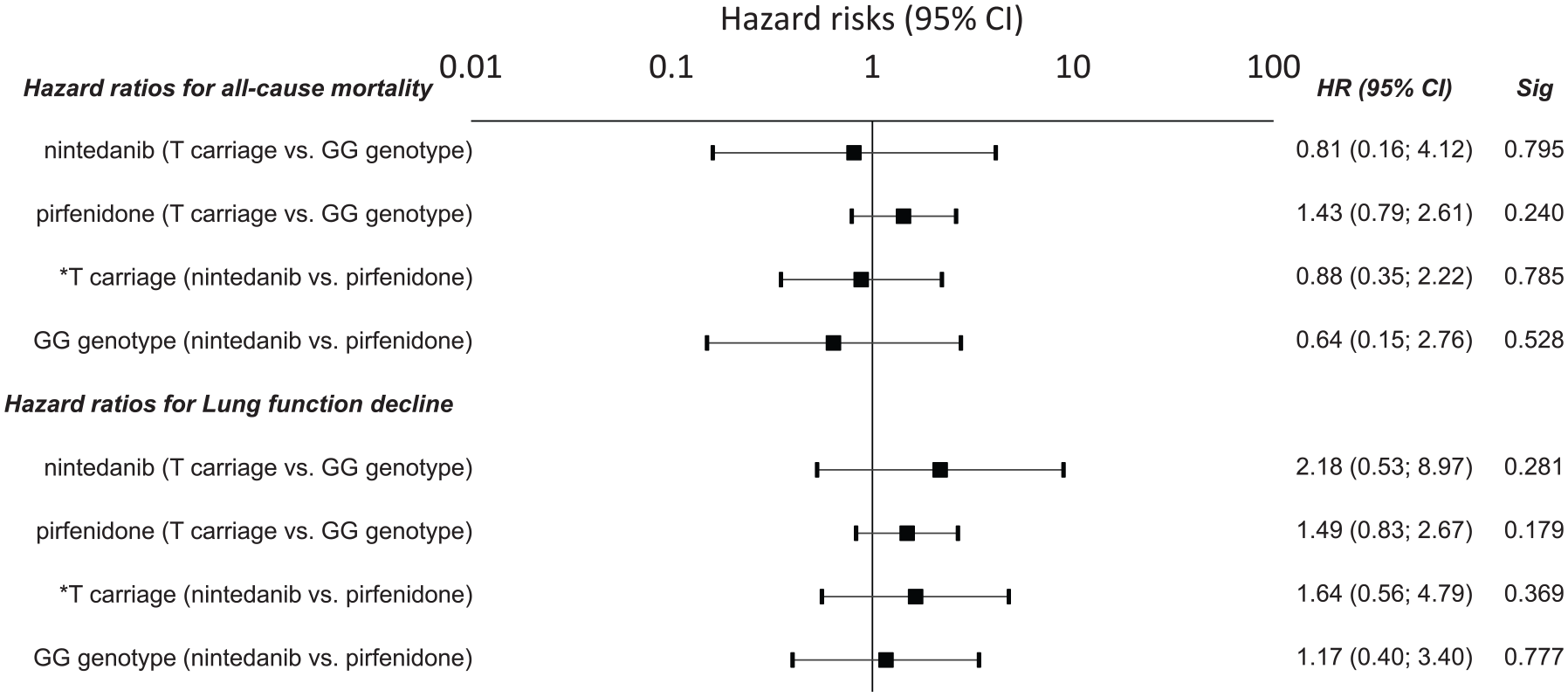
Hazard risk for all-cause death and a lung function decline in IPF patients with respect to the MUC5B rs35705950. Hazard risks (HRs) are presented with 95% confidence intervals noted in parentheses.
Influence of DSP variants on IPF prognosis and antifibrotic treatment outcome
Mortality in IPF patients with DSP rs2076295 G* allele treated with antifibrotic agents (pirfenidone or nintedanib) was lower compared with these without antifibrotic treatment (p = 0.016) (Figure 4). After stratifying by DSP rs2076295, the Kaplan–Meier curve showed that patients with the *G allele benefitted from treatment with nintedanib (p = 0.004) but not pirfenidone (p = 0.612) compared with those with a TT genotype (Figure 5). Patients with a TT genotype (rs2076295), however, showed better performance in terms of OS on pirfenidone (p = 0.033) than on nintedanib. For *G allele carriage, the difference in mortality between treatment with nintedanib and pirfenidone did not reach significance (p = 0.110) (Figure 5).

Mortality in IPF patients subdivided according to the carriage of DSP rs2076295*G allele treated with (a) nintedanib or pirfenidone or (b) without antifibrotic treatment and comparison between nintedanib or pirfenidone treatments versus without antifibrotic treatment for (c) *T allele carriers and (d) GG genotypes.

Mortality in IPF patients subdivided according to the carriage of DSP rs2076295*G allele treated with (a) nintedanib and (b) pirfenidone and comparison between nintedanib and pirfenidone treatments for (c) *G allele carriers and (d) TT genotypes.
Carriers of the DSP rs2076295*G allele benefitted from treatment with nintedanib, compared with patients with TT genotypes who had a shorter OS (HR = 7.99; 95% CI = 1.56–40.90; p = 0.013) and a faster decline in lung function (HR = 8.51; 95% CI = 1.68–43.14; p = 0.010) (Figure 6). The observed results from nintedanib treatment were confirmed by cross-validation, showing a mean HR ± SD = 6.59 ± 3.14 for the risk of death and HR ± SD = 9.56 ± 4.3 for a decline in lung function. There was also a trend that patients with the DSP rs2076295*G allele benefitted more from treatment with nintedanib than pirfenidone, as they showed a faster decline in lung function on pirfenidone (p = 0.059; HR = 4.04; 95% CI = 0.95–17.10). Patients with a TT genotype, however, had a longer OS (p = 0.040; HR = 0.35; 95% CI = 0.13–0.95) but not a slower decrease in lung function (p = 0.394; HR = 0.72; 95% CI = 0.18–3.73) when treated with pirfenidone rather than nintedanib (Figure 6).

Hazard risk for all-cause death and a lung function decline in IPF patients with respect to the DSP rs2076295 variants. Hazard risks (HRs) are presented with 95% confidence intervals noted in parentheses.
Outcome prediction: univariate and multivariate analyses
To investigate the association of clinical and demographic factors and lung function parameters with survival and disease outcome, univariate and multivariate analysis by using Cox regression was performed. In the univariate analysis, male gender, clubbing finger, cough, low FVC and low DLCO at the time of diagnosis were found as risk predictors of death. In the multivariate model, smoking, older age at the time of IPF diagnosis, FVC and DLCO at the IPF diagnosis were associated with survival. There was no association between MUC5B rs35705950 T* allele and DSP rs2076295 G* allele carriers and survival nor acute exacerbations in the IPF cohort (Table 4).
Association between exacerbation, IPF treatment and studied variants.

IPF, idiopathic pulmonary fibrosis.
Discussion
This study investigated the treatment outcome in IPF patients treated with antifibrotic drugs nintedanib and pirfenidone. The results showed that both antifibrotic agents have a positive impact on mortality, particularly in DSP rs2076295 G* and MUC5B rs35705950 T* allele carriers. In addition, the DSP rs2076295*G allele has a positive impact on OS and lung function decline in IPF patients when treated with nintedanib but not pirfenidone.
We studied the association of two common polymorphisms with nintedanib and pirfenidone treatment. These are the only two antifibrotic drugs currently approved for IPF treatment. Both drugs act through different mechanisms.21,22 Nintedanib is a tyrosine kinase inhibitor that also targets vascular endothelial growth factor receptors (VEGFR 1–3), fibroblast growth factor receptors (FGFR 1–3) and platelet-derived growth factor receptors (PDGFR).23,24 Regarding pirfenidone, in vitro- studies of normal human lung fibroblasts showed that pirfenidone abrogates TGF-β1-stimulated collagen synthesis and reduces fibroblast proliferation, production of alphasmooth muscle actin (α-SMA) induced by TGF-β, and the level of procollagen. 25 A recent study on mice of bleomycin-induced pulmonary fibrosis showed that pirfenidone is more effective in the prophylactic fibrosis model, while nintedanib is more effective in early and late treatment models. 22 Pharmacogenetic data on the use of both antifibrotics in the treatment of IPF are limited.
We first analysed the impact of the common promoter variant in gene MUC5B (rs35705950), which is the strongest risk factor for the development of idiopathic interstitial pneumonias and IPF.7,8,26,27 MUC5B polymorphism affects the rheological properties of airway mucus, mucociliary transport and is associated with a more typical subpleural distribution of fibrosis and with a greater proportion of confident radiological diagnosis [probable UIP (usual interstitial pneumonia) and UIP]. 8 The risk T allele occurs in 31–42% of patients with IPF;7,26,28,29 in our real-world IPF cohort, this variant was detected in 55% of patients. Carriage of the risk T allele is associated with elevated expression of MUC5B in the pseudostratified mucociliary epithelium, better prognosis and improved survival.9,30 Animal models have proved that improved survival might be linked to better microbial host defence through enhanced mucin production.28,31 Our analysis revealed decreased mortality in IPF patients with MUC5B rs35705950 T* allele treated with antifibrotic agents (nintedanib or pirfenidone) compared with a group without antifibrotic treatment. When the subanalysis in patients treated with nintedanib or pirfenidone was performed, no difference in treatment outcome or lung function decline with regard to T allele (rs35705950) carriage or genotype was detected. Recent study on 88 IPF patients (69% T allele carriers) on antifibrotic therapy (pirfenidone or nintedanib) also observed association of the MUC5B *T allele with longer survival compared with a GG genotype, but not with lung function decline. 32 Other recent study investigated 62 IPF patients on antifibrotic treatment for MUC5B rs35705950 and TOLLIP rs5743890 polymorphisms. 33 This study reported that minor allele C in TOLLIP, an inhibitor of the toll-like receptors (TLRs) 2 and 4, but not MUC5B, was associated with worse survival, acute exacerbation and disease progression. 33 Similarly, in our cohort no association of MUC5B rs35705950 with acute exacerbation IPF nor radiological findings were observed. Other study has shown that the MUC5B variant does not affect oral N-acetylcysteine treatment in IPF. 5 The role of the MUC5B *T allele in pharmacogenomics should be further investigated.
Next, we investigated the association of another common polymorphism (rs2076295) in the DSP gene that is associated with IPF 10 on the outcome of treatment with nintedanib and pirfenidone. Desmoplakin, a protein encoded by the DSP gene, is vital for cell contacts, wound repair and epithelial barrier function.7,10,11 Its expression is increased in the lungs of IPF patients, predominantly in the airway epithelium.7,11 The DSP *G allele (rs2076295), present in 80.5% of IPF patients, is associated with a decreased gene expression in both the proximal and distal airways in a dose-dependent manner, even in IPF-affected lungs. 10 In our cohort, the presence of DSP G* allele was associated with a radiological diagnosis and an interstitial score. The association of DSP polymorphism and radiological patterns on HRCT of thorax has not been reported yet and deserves future investigations. Our data showed that IPF patients who carry the DSP *G allele (rs2076295) (GG and GT genotypes) have lower mortality and a slower lung function decline than those with TT genotypes when treated with nintedanib. Importantly, there was a trend of slower lung function decline in DSP *G allele (rs2076295) carriers when treated with nintedanib rather than pirfenidone. It may therefore be suggested that lower expression of DSP in the airway epithelium, caused by the rs2076295*G allele, may contribute to increased efficiency of nintedanib in IPF patients, particularly for those with a GT genotype. Recently, it has been reported that nintedanib, but not pirfenidone, has an effect on the pulmonary epithelium, restoring the expression of epithelial genes as well as increasing proSP-C protein expression and affecting SP-C secretion in human lung tissue cultures of pulmonary fibrosis patients. 34 Because desmoplakin is highly expressed in the epithelium, our data point to the role of the lung epithelium not only in the pathogenesis of IPF but also in the effectiveness of antifibrotic drugs. Our data, however, also showed that IPF patients with a TT genotype (19.5%) could benefit from pirfenidone treatment, resulting in longer OS than when treated with nintedanib. A recent study revealed that the T allele is associated with longitudinal change in quantitative emphysema in patients with chronic obstructive pulmonary disease. 35 Emphysema is also highly prevalent in smokers with IPF, coexisting as a combined pulmonary fibrosis emphysema36,37 that has a different lung physiology and clinical outcomes than IPF alone. 38 How the DSP *T allele (rs2076295) affects the antifibrotic treatment response and its association with emphysema in IPF, however, are still unknown and deserve future research.
We also evaluated the association of clinical, demographic factors and lung function parameters with survival and disease outcome in univariate and multivariate analyses. As in the previous EMPIRE registry data analysis, we confirmed association of favourable prognosis in IPF with female sex, younger age, greater predicted FVC and greater predicted DLCO at diagnosis together with cough and antifibrotic treatment. 39 In the multivariate model, smoking, age at the time of IPF diagnosis, FVC and DLCO at the IPF diagnosis were associated with survival. Our analysis also revealed that MUC5B rs35705950 T* and DSP rs2076295 G* alleles do not affect survival in IPF as a whole, but contribute to better outcome on antifibrotic drugs.
Our study has several limitations. We included a modest cohort of patients with IPF in the study due to the short follow-up time on antifibrotics of other patients on the registry. In addition, our data from a single registry should be verified with an independent group of IPF patients of different ethnicities. Despite these limitations, our exploratory study on a similarly sized group of patients as another IPF pharmacogenomic studies5,32,33 demonstrated an association between the profibrotic variant and antifibrotic treatment outcomes.
Conclusion
Taken together, our real-world study showed that IPF patients with MUC5B T* allele or DSP G* allele profit from antifibrotic treatment (nintedanib or pirfenidone) by longer survival. In addition, our pilot study revealed that carriers of the DSP rs2076295*G allele benefit from treatment with nintedanib, compared with IPF patients with a TT genotype who had a shorter OS and a faster decline in lung function on this drug and benefit from pirfenidone treatment. MUC5B rs35705950 did not impact the outcome of treatment with either nintedanib or pirfenidone. Our study has highlighted the contribution of pharmacogenetics to treatment outcomes and may help to identify genetic subgroups likely to benefit from a particular antifibrotic therapy, as desired in the era of precision medicine.
Footnotes
Acknowledgements
MaD and EK contributed equally to this work.
Author contributions
MaD, EK and MiD designed the study, interpreted the data and drafted the manuscript; MV critically revised the manuscript; SL and PS performed the statistical data analysis; PS and EK performed the genetic analysis; MaD, MS, VB, MP, MZ, RB, VL, LS, PL, HS, JP, TS and PM collected the patient characteristics and contributed to the data interpretation. All authors have read and approved the final manuscript and agreed to be accountable for all aspects of the work.
Conflict of interest statement
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by Internal agency of Palacky University (Grant No. IGA_LF_2021_015), Ministry of Health of Czech Republic (MH CZ – DRO FNOL, 00098892) and Czech Pneumological and Phthiseological Society (publication fee grant).