
Abstract
Close monitoring of patients with fibrosing interstitial lung diseases (ILDs) is important to enable prompt identification and management of progressive disease. Monitoring should involve regular assessment of physiology (including pulmonary function tests), symptoms, and, when appropriate, high-resolution computed tomography. The management of patients with fibrosing ILDs requires a multidisciplinary approach and should be individualized based on factors such as disease severity, evidence of progression, risk factors for progression, comorbidities, and the preferences of the patient. In this narrative review, we discuss how patients with fibrosing ILDs can be effectively monitored and managed in clinical practice.
Introduction
Interstitial lung diseases (ILDs) are a large and heterogeneous group of disorders, which in some cases become fibrotic and progressive. Idiopathic pulmonary fibrosis (IPF) is relentlessly progressive 1 and may be regarded as the ‘prototypic’ progressive fibrosing ILD. 2 Other ILDs, such as fibrotic hypersensitivity pneumonitis (HP) and those associated with autoimmune diseases, may also develop progressive fibrosing disease behavior characterized by worsening respiratory symptoms and hypoxemia, decline in lung function, increasing fibrotic abnormalities on high-resolution computed tomography (HRCT), and early mortality.2,3
Diagnosis of fibrosing ILDs
A differential diagnosis of ILD is made based on a comprehensive medical history, clinical examination, serologies, HRCT, and, if needed, bronchoalveolar lavage or lung biopsy.1,4,5 Multidisciplinary discussion, involving a pulmonologist, a radiologist, and, where appropriate, a rheumatologist and pathologist, is regarded as the gold standard for differential diagnosis of ILD.4,6 This approach may be particularly valuable in the diagnosis of autoimmune disease-related ILDs.7,8 Some patients do not fulfill the criteria for a specific ILD diagnosis even after multidisciplinary discussion; in such cases, a ‘working diagnosis’ may be appropriate but should be re-assessed regularly. 4 Patients with interstitial lung abnormalities (ILAs) that do not meet criteria for diagnosis of ILD and who have risk factors for progression to ILD such as smoking or other inhalational exposures, or evidence of fibrosis, should be monitored closely to ensure that progression to ILD is detected promptly. 9
Diagnostic criteria for IPF developed by the Fleischner Society 4 and jointly by the American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society 1 proposed similar categories to interpret HRCT scans in patients suspected of having ILD. The categories proposed by the Fleischner Society are: typical usual interstitial pneumonia (UIP) CT pattern; probable UIP CT pattern; CT pattern indeterminate for UIP; CT features most consistent with a non-IPF diagnosis. 4 A typical UIP pattern is defined by a peripheral and basal distribution of reticulation, architectural distortion, traction bronchiectasis, and subpleural honeycombing (Figure 1). A probable UIP pattern has similar features but without honeycombing. Typical and probable UIP patterns usually preclude biopsy in patients suspected of having IPF. A pattern indeterminate for UIP has some inconspicuous features suggestive of a non-UIP pattern such as a lack of an apical-basilar gradient or scattered mosaic attenuation. Features most consistent with a non-IPF diagnosis include atypical distributions such as mid- or upper-lung zone predominant fibrosis or peribronchovascular predominance with subpleural sparing. Additional features that suggest an alternative diagnosis include extensive ground-glass opacities (without acute exacerbation), extensive mosaic attenuation with sharply defined lobular air trapping on expiration involving three or more lobes, diffuse nodules or cysts. Certain patterns of fibrosis on HRCT are suggestive of specific diagnoses. Non-specific interstitial pneumonia (NSIP) typically manifests with peripheral ground-glass opacities that often demonstrate subpleural sparing (Figure 2(a)). Certain myositis syndromes such as the anti-synthetase syndrome often manifest with a combined NSIP/organizing pneumonia (OP) pattern (Figure 2(b)). The fibrotic form of HP should be considered with the ‘three-density pattern’ (i.e. normal lung, hyperlucent lung and ground-glass opacities), mid- and upper-lung zone distribution of fibrosis with ground-glass opacities, mosaic lung attenuation and expiratory air trapping (Figure 2(c) and (d)). 5

Typical usual interstitial pneumonia (UIP) pattern on high-resolution computed tomography (HRCT) of the chest in a 77-year-old man with idiopathic pulmonary fibrosis. (a) Axial and coronal (b) HRCT images show peripheral and basal predominant reticulation, architectural distortion, traction bronchiectasis (white arrowhead) and subpleural honeycombing (black arrow).

Patterns of fibrosis on high-resolution computed tomography (HRCT) scans of the chest suggestive of specific diagnoses. (a) Non-specific interstitial pneumonia (NSIP) in a 65-year-old man; axial HRCT image shows peripheral predominant ground-glass opacities with areas of subpleural sparing (arrows) typical of NSIP. (b) Anti-synthetase syndrome in a 52-year-old woman; axial HRCT image shows lower lobe peribronchovascular consolidation with traction bronchiectasis typical of a combined NSIP/organizing pneumonia (OP) pattern often seen with a myositis syndrome. Note areas of subpleural sparing in the left lower lobe. (c and d) Fibrotic hypersensitivity pneumonitis secondary to bird antigen exposure in a 54-year-old man; axial (c) and coronal (d) HRCT images show mid- and upper-lung zone and peribronchovascular predominant fibrosis with traction bronchiectasis (arrow), mosaic attenuation (arrowhead) and areas of ground-glass opacity. Note three different lung densities in keeping with the ‘three-density pattern’ or headcheese sign.
Progression of fibrosing ILDs
IPF is always progressive, but its rate of progression varies among individuals and acute exacerbations of the disease are unpredictable and often fatal.1,10 Progressive disease behavior is also observed in a subgroup of patients with other fibrosing ILDs including fibrotic HP;5,11 ILDs associated with autoimmune diseases such as systemic sclerosis,12,13 rheumatoid arthritis,14,15 and anti-synthetase myositis; 16 sarcoidosis-related ILD; 17 exposure-related ILDs; 18 and unclassifiable ILD. 19
When a typical UIP-fibrotic pattern is present on HRCT, decline in lung function in patients with other ILDs may be as rapid as in patients with IPF.3,19–24 As well as decline in lung function, progression of ILD may manifest as an increase in the extent of fibrotic abnormalities on HRCT12,25,26 or as deterioration in symptoms, exercise capacity, or oxygen saturation during exercise, and quality of life.27–30
Although the course of fibrosing ILD is difficult to predict, observational studies have identified a number of factors associated with mortality. A greater extent of fibrosis on HRCT has been associated with mortality in studies across ILDs.12,21,26,31–35 In an analysis of 519 patients with systemic sclerosis from a nationwide Norwegian cohort, the standardized mortality ratio over a mean observation period of 10 years increased from approximately 2 in patients with no fibrosis on HRCT to 5 in patients with an extent of fibrosis > 10% and 8 in those with an extent of fibrosis > 25%. 12 In an analysis of data from 159 patients with RA-ILD, patients with an extent of fibrosis ⩾ 20% on HRCT had over twice the risk of dying during a 14-year follow-up period than patients with a lesser extent of fibrosis. 31 In patients with HP, the predominant presence of fibrosis on HRCT or lung biopsy or both is the strongest predictor of mortality and led to the re-categorization of HP into fibrotic and non-fibrotic subtypes in the latest international diagnostic guidelines. 5
A decline in forced vital capacity (FVC) of ⩾ 10% of the predicted value has consistently been associated with mortality in patients with ILDs.3,24,34,36–38 Among 331 patients with progressive fibrosing ILDs who received placebo in the INBUILD trial, a relative decline in FVC ⩾ 10% predicted was associated with a more than three-fold increase in the risk of death over 52 weeks. 24 Among 137 patients with RA-ILD at a US center, patients with an absolute decline in FVC ⩾ 10% predicted at any time over a median follow-up of 4.8 years had a 2.5-fold greater death rate than patients without such a decline. 36 An analysis of 112 patients with chronic HP found that median survival was 53 months in patients who had an absolute decline in FVC ⩾ 10% predicted 6–12 months after diagnosis, compared with 139 months among those who did not. 38
Factors specific to particular ILDs should also be considered. In patients with HP, lower lymphocyte count in bronchoalveolar lavage fluid 39 and continued exposure to an inciting antigen 40 are associated with worse outcome. In patients with autoimmune disease-related ILDs, several autoantibodies have been associated with a greater risk of progression of ILD; for example, anti-topoisomerase I (Scl-70) antibody in patients with early systemic sclerosis 41 and anti-cyclic citrullinated peptide antibody in patients with rheumatoid arthritis. 35 In patients with antisynthetase myositis-ILD, negativity for anti-Jo-1 antibodies is associated with higher mortality. 42
Monitoring of fibrosing ILDs
Close monitoring of patients with ILDs is important to assess disease progression and inform patient care, such as initiation of pharmacotherapy and counseling. Monitoring should involve regular assessment of symptoms, physiology (e.g. pulmonary function and six-minute walk testing) and, where appropriate, HRCT. International guidelines suggest that patients with IPF should have FVC and diffusing capacity of the lung for carbon monoxide (DLco) measured every 3 to 6 months (or sooner if clinically indicated). 43 The 6-minute walk test can be a useful tool to assess disease progression in patients with IPF, 44 but may be more challenging to perform in patients with rheumatic diseases due to the impact of extra-pulmonary problems affecting ambulation such as joint pain and fatigue. In addition, evaluation for oxygen desaturation and need for supplemental oxygen during a walk test in patients with Raynaud’s phenomenon may be confounded by falsely low peripheral oxygen saturation readings (SpO2) due to reduced digital perfusion. In such patients, it may be useful to perform 6MWTs using both ear and finger pulse oximetry. Monitoring changes in symptoms may be based on a simple overall assessment, but questionnaires can also be used to provide a more objective measure of disease progression. 45 When interpreting patient-reported outcomes in patients with ILDs, it should be considered that deterioration in symptoms, exercise capacity, or quality of life may reflect deterioration in manifestations of disease other than ILD, or new or worsening comorbidities such as pulmonary hypertension. Experts in the management of patients with SSc-ILD have proposed that patients should be monitored for disease progression every 3–6 months using multiple methods, including symptom assessment, pulmonary function tests, exercise-induced blood oxygen desaturation and, where appropriate, HRCT.46–48 However, for most ILDs, there are no guidelines for how patients should be monitored. An individualized approach is essential, taking into account factors such as disease severity, evidence of progression, risk factors for progression, comorbidities, quality of life, and the patient’s preferences (Figure 3). Regular monitoring of patients with ILDs also provides an opportunity to screen for onset or deterioration of other disease manifestations and comorbidities. Imaging of patients with ILD and deteriorating respiratory symptoms may reveal problems other than progression of ILD such as lung carcinoma 49 (Figure 4).

Proposed approach to monitoring and management of non-IPF fibrosing ILDs.

High-resolution computed tomography scans of the chest from a 68-year-old man with idiopathic pulmonary fibrosis and a right upper lobe lung cancer. Coronal images obtained in 2015 (left), 2018 (middle) and 2020 (right) show an enlarging subpleural nodule (arrow) in the right upper lobe proven to represent a primary lung cancer. Note peripheral right lower lobe reticulation and honeycombing in keeping with a usual interstitial pneumonia (UIP) pattern of fibrosis.
There are no consensus recommendations regarding imaging follow-up for patients with progressive fibrosing ILDs. Many providers will obtain an HRCT at the patient’s initial presentation and then every 12–18 months to assess for progression. There are no data to support the use of chest radiography over HRCT for follow-up of patients with confirmed ILD. More frequent imaging is often obtained when patients present with acute or worsening symptoms or declining pulmonary function tests.
Management of fibrosing ILDs
IPF is an inexorably progressive disease. As such, barring any contraindications, patients with IPF should be offered prompt treatment with an approved antifibrotic therapy (nintedanib or pirfenidone) to slow the progression of their disease. 50 Immunosuppression should not be used for chronic treatment of IPF 51 and there is evidence that it may be harmful. 52 As IPF progresses, action should be taken to optimize symptom relief and preserve quality of life, including referral for palliative care as appropriate. 53 For fibrosing ILDs other than IPF, the optimal sequence, combination and timing of use of immunosuppressants, nintedanib, and supportive therapies has not been established. Management of these diseases requires a multidisciplinary and individualized approach that takes into account the severity of ILD, evidence of progression, other disease manifestations and comorbidities, as well as the patient’s preferences46,54–56 (Figure 3). For exposure-related ILDs, such as HP, drug-induced ILD, and occupational ILDs/pneumoconioses, it is essential to identify, eliminate, and avoid culprit exposures.
Based on data from the INPULSIS, SENSCIS and INBUILD trials (Table 1), nintedanib 150 mg twice daily (or 100 mg twice daily for patients with mild hepatic impairment) has been approved by regulatory authorities including the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of IPF, for slowing decline in FVC in patients with ILD associated with systemic sclerosis, and for the treatment of chronic fibrosing ILDs with a progressive phenotype. Although the INBUILD trial was not designed or powered to evaluate individual ILDs, subgroup analyses suggested that there was no heterogeneity in the rate of decline in FVC in the placebo group 24 or in the treatment effect of nintedanib across subgroups by ILD diagnosis 62 (Figure 5). Further, a comparison of data from the placebo groups of the INPULSIS and INBUILD trials showed that the rate of decline in FVC over 52 weeks was similar in patients with IPF as in patients with other fibrosing ILDs and a UIP-like fibrotic pattern on HRCT (mean −223.2 versus −214.6 mL/year). 24 Across all clinical trials, the adverse events associated with nintedanib were predominantly gastrointestinal events, particularly diarrhea.59,63,64 Diarrhea should be managed promptly with adequate hydration and antidiarrheal agent (e.g. loperamide), and with dose reduction from 150 mg twice daily to 100 mg twice daily or treatment interruption if symptoms persist. 65 Liver enzymes and bilirubin should be monitored prior to the initiation of nintedanib, at regular intervals during the first 3 months of treatment, and then periodically or as clinically indicated. 65
Randomized placebo-controlled phase III trials of FDA-approved treatments for ILDs.

FDA, US Food and Drug Administration; FVC, forced vital capacity; HRCT, high-resolution computed tomography; ILD, interstitial lung diseases; IPF, idiopathic pulmonary fibrosis; 6MWT, 6-minute walk test distance; SSc, systemic sclerosis.
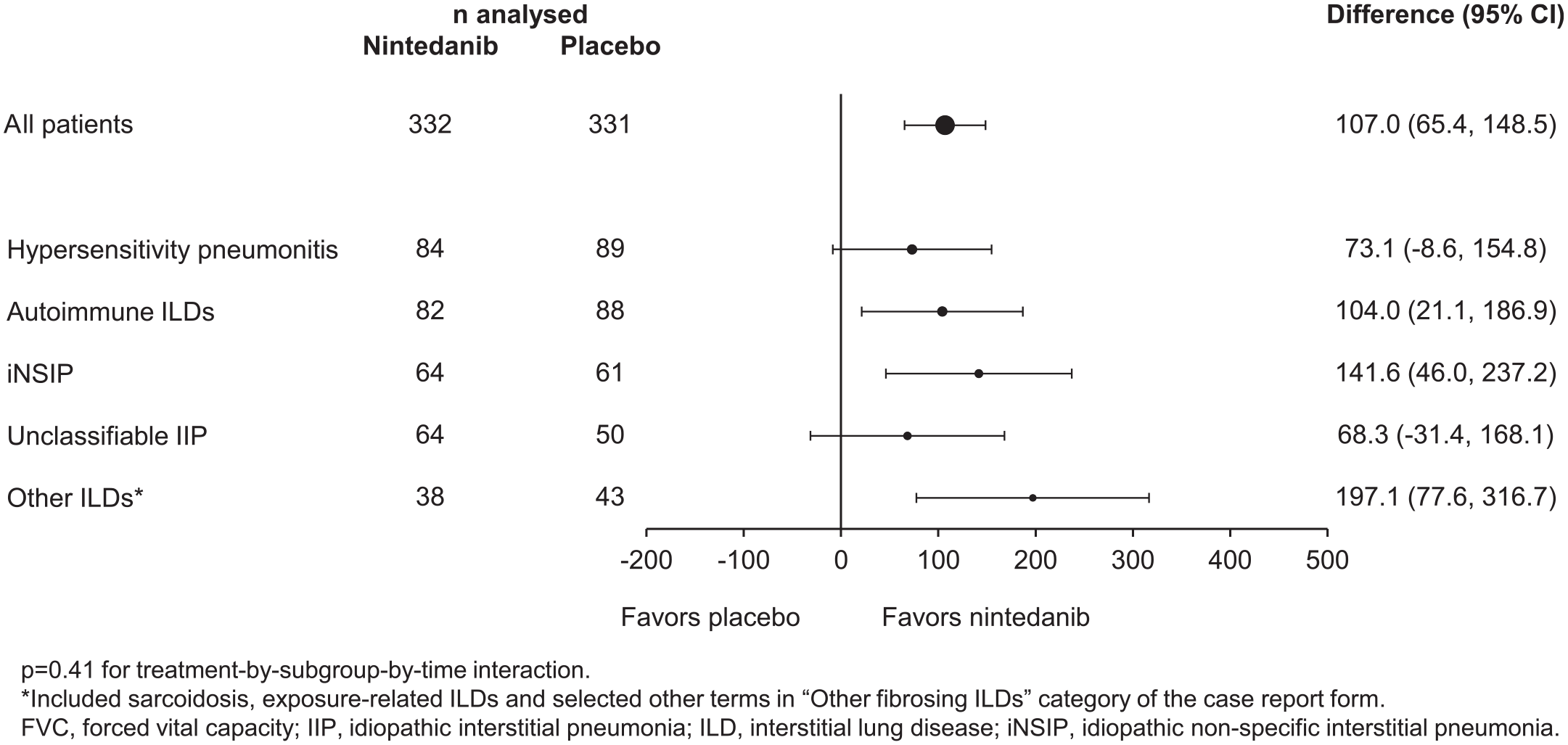
Subgroup analysis of treatment effect of nintedanib versus placebo on rate of decline in FVC (mL/year) over 52 weeks in patients with progressive fibrosing ILDs other than IPF in the INBUILD trial. Reprinted from Wells and colleagues, 62 Copyright (2020), with permission from Elsevier.
Pirfenidone 801 mg three times daily has been approved for the treatment of IPF, based on demonstration of a reduced rate of decline in FVC compared with placebo in the ASCEND trial 61 (Table 1). A randomized, placebo-controlled trial of pirfenidone in 253 patients with progressive fibrosing unclassifiable ILD failed to meet its primary endpoint of change in FVC % predicted over 24 weeks measured by daily home spirometry, but in-clinic spirometry suggested that pirfenidone reduced the rate of decline in FVC, with a mean decline of −17.8 mL in the pirfenidone group compared to −113.0 mL in the placebo group. 19 Commonly reportedly side-effects associated with pirfenidone included gastrointestinal disorders, photosensitivity and rash.19,61,60 Patients are advised to avoid exposure to sunlight and to wear protective clothing and sunscreen. Adverse events can be managed by dose reductions or treatment interruptions. 66 Liver enzymes and bilirubin should be monitored prior to the initiation of pirfenidone, monthly for the first 6 months, and every 3 months thereafter. 66 No clinical trials assessing the efficacy of pirfenidone in treating ILDs other than IPF and unclassifiable ILD have been completed but several are in progress (NCT03260556, NCT02808871, NCT03856853).
In patients with IPF, treatment with nintedanib plus add-on pirfenidone or pirfenidone plus add-on nintedanib over 24 weeks demonstrated a safety and tolerability profile in line with the adverse event profiles of each drug.67,68 Few data are available on switching between antifibrotic therapies. Among 782 patients in a large US registry, most patients taking an antifibrotic therapy at enrollment were still taking the same therapy approximately 6 months later. 69
Most patients with autoimmune diseases are receiving immunosuppressant therapy to treat their systemic disease irrespective of whether they have ILD. There is evidence from randomized, double-blind, controlled trials to support the use of mycophenolate mofetil and cyclophosphamide in the treatment of SSc-ILD70–72 and the use of these therapies has been recommended by experts in the field.46–48 No other randomized, double-blind controlled trials have demonstrated a benefit of immunosuppressant therapy in the treatment of autoimmune disease-associated ILDs, but they are widely used, 73 likely based on extrapolation from findings in patients with SSc-ILD and the findings of uncontrolled studies. High RA disease activity has been associated with increased risk of developing RA-ILD, 74 suggesting that treatment of underlying rheumatic disease may also influence ILD risk. Immunosuppressants are also frequently used to treat fibrotic HP, but in the absence of well-designed prospective randomized controlled trials, it remains unclear whether they are effective in slowing its progression.75,76
At present, neither expert consensus nor clinical practice guidelines exist for specific pharmacologic treatment of progressive fibrosing ILDs. If the decision is made to start treatment, we believe that an initial ILD diagnosis established by a multidisciplinary discussion should inform the initial treatment choice (e.g. antifibrotics for IPF, immunosuppression for CTD-ILDs and non-fibrotic HP). If immunosuppression is contraindicated, not tolerated, or declined due to patient preference, nintedanib (the only antifibrotic drug currently approved for non-IPF ILD indications) should be considered. Of note, high-quality data are lacking regarding optimal first-line treatment for various non-IPF ILDs. For patients with progressive fibrosing ILD, as defined by the INBUILD trial criteria, 59 despite or in lieu of immunosuppression (e.g. due to recurrent infection), nintedanib may be a reasonable choice as monotherapy or as add-on combination therapy with immunosuppressive agents.
Patients with fibrosing ILDs may benefit from non-pharmacological therapies such as smoking cessation, pulmonary rehabilitation, exercise training, and attendance at patient support groups (Figure 6).77,78 An official clinical practice guideline recently published by the ATS provided strong recommendations for long-term oxygen use in patients with ILD and severe chronic resting hypoxemia and conditional recommendations for oxygen use in patients with ILD and severe exertional hypoxemia, while recognizing that patients and their caregivers need education on how to use oxygen equipment. 79

Beyond pharmacotherapy: a holistic approach to care for patients with fibrosing interstitial lung diseases (ILDs).
The treatment of patients with fibrosing ILDs should be re-assessed if symptoms or lung function deteriorate, ideally based on multidisciplinary discussion. Select patients with rapidly progressive SSc at risk of organ failure may be considered for autologous hematopoietic stem cell transplantation at specialized centers, based on a careful assessment of the potential risks-benefits for the individual. 80 Lung transplantation should be considered for patients with IPF at an early stage of disease. 43 A consensus statement from the International Society for Heart and Lung Transplantation (ISHLT) specified that referral for lung transplantation should be based on predictors of poor prognosis such as histopathologic or radiographic evidence of UIP or fibrosing NSIP, impaired lung function (FVC < 80% predicted, DLCO < 40% predicted), or supplemental oxygen use. The timing of listing for transplantation should be based on factors indicating physiological decline such as a decline in FVC ⩾ 10% predicted, a decline in DLco ⩾ 15% predicted over 6 months, or hospitalization due to respiratory decline. 81 Similar guidance was provided for patients with autoimmune disease-associated ILD who have not responded to treatment and do not have extrapulmonary contraindications to transplantation. 81 Survival rates after lung transplant appear to be similar between patients with IPF and patients with autoimmune disease-associated ILDs82,83 or sarcoidosis. 84 Survival rates after lung transplant among patients with HP may be greater 85 but more data are needed.
Conclusions
Fibrosing ILDs have an unpredictable clinical course. A proportion of patients with fibrosing ILDs develop a progressive phenotype characterized by worsening symptoms, declining exercise capacity, increasing hypoxemia, decline in lung function, increasing fibrotic abnormalities on HRCT, and early mortality. Patients with fibrosing ILDs require close monitoring to ensure that disease progression is identified promptly. The management of patients with fibrosing ILDs requires a multidisciplinary and individualized approach.
Supplemental Material
sj-docx-1-tar-10.1177_17534666211039771 – Supplemental material for Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians
Supplemental material, sj-docx-1-tar-10.1177_17534666211039771 for Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians by Anoop M. Nambiar, Christopher M. Walker and Jeffrey A. Sparks in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-2-tar-10.1177_17534666211039771 – Supplemental material for Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians
Supplemental material, sj-docx-2-tar-10.1177_17534666211039771 for Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians by Anoop M. Nambiar, Christopher M. Walker and Jeffrey A. Sparks in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-docx-3-tar-10.1177_17534666211039771 – Supplemental material for Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians
Supplemental material, sj-docx-3-tar-10.1177_17534666211039771 for Monitoring and management of fibrosing interstitial lung diseases: a narrative review for practicing clinicians by Anoop M. Nambiar, Christopher M. Walker and Jeffrey A. Sparks in Therapeutic Advances in Respiratory Disease
Footnotes
Acknowledgements
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). Editorial support was provided by Elizabeth Ng and Wendy Morris of FleishmanHillard Fishburn, London, UK, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc (BIPI). Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Unrelated to this work, AMN has received federal research support from San Antonio Claude D. Pepper Older Americans Independence Center’s (OAIC) Pilot/Exploratory Studies Core (PESC) Grant; non-profit research support from the Pulmonary Fibrosis Foundation; industry research grants from Boehringer Ingelheim, Galapagos, Nitto Denko, and FibroGen; and consultancy and speaker’s bureau fees from Boehringer Ingelheim, Roche/Genentech, and Veracyte. CMW discloses royalties from Elsevier and Amirsys and is part of the speaker’s bureau for Boehringer Ingelheim. JAS is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases (grant numbers K23 AR069688, R03 AR075886, L30 AR066953, P30 AR070253, P30 AR072577), the Rheumatology Research Foundation (R Bridge Award), and the R. Bruce and Joan M. Mickey Research Scholar Fund. Dr. Sparks has received research support from Bristol-Myers Squibb and performed consultancy for Bristol-Myers Squibb, Gilead, Inova Diagnostics, Optum, and Pfizer unrelated to this work.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. If the article is accepted, page processing/open access charges would be paid for by Boehringer Ingelheim Pharmaceuticals, Inc. The authors did not receive any payment for development of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.