
Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive and fatal lung disease in which most patients die within 3 years of diagnosis. With an unknown etiology, IPF results in progressive fibrosis of the lung parenchyma, diminishing normal lung function, which results in respiratory failure, and eventually, death. While few therapies are available to reduce disease progression, patients continue to advance toward respiratory failure, leaving lung transplantation the only viable option for survival. As incidence and mortality rates steadily increase, the need for novel therapeutics is imperative. The receptor for advanced glycation endproducts (RAGE) is most highly expressed in the lungs and plays a significant role in a number of chronic lung diseases. RAGE has long been linked to IPF; however, confounding data from both human and experimental studies have left an incomplete and perplexing story. This review examines the present understanding of the role of RAGE in human and experimental models of IPF, drawing parallels to recent advances in RAGE biology. Moreover, this review discusses the role of RAGE in lung injury response, type 2 immunity, and cellular senescence, and how such mechanisms may relate to RAGE as both a biomarker of disease progression and potential therapeutic target in IPF.
The reviews of this paper are available via the supplemental material section.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive and universally fatal disease in which individuals usually die within 3 years of diagnosis due to progressive fibrosis of the pulmonary parenchyma. 1 Pathologically, IPF demonstrates a pattern of fibrosis referred to as usual interstitial pneumonia (UIP). UIP is recognized microscopically by the presence of temporally heterogeneous pulmonary fibrosis in which there are areas of mature fibrosis, immature fibrosis, and histologically normal alveolar parenchyma, sometimes in the same high-power field of view. The fibrosis is generally more severe in the lower lobes, lung bases and subpleural regions, but the disease progresses to involve more central areas with time. 2 This irreversible destruction of the normal lung architecture significantly reduces lung function, leading to respiratory failure. While there are known causes of pulmonary fibrosis that can lead to UIP such as collagen vascular disorders or asbestosis, most causes of UIP have no known cause and are diagnosed as IPF. 3
Although antifibrotic therapies such as nintedanib and pirfenidone have shown promise in slowing disease advancement, patients continue to progress toward respiratory failure, leaving lung transplantation the only other option to promote survival and avoid death.4,5 Therefore, there is a vital need for effective biomarkers to enhance early diagnosis and novel therapeutics to halt disease progression, improve quality of life and survival rates.
Tissue repair is an essential biological function in response to damage caused by endogenous or exogenous insults, in which damaged cells are removed, parenchymal space is repaired and structural cells reorganized to restore normal tissue function. 6 However, when dysregulated, this can lead to excessive remodeling and deposition of extracellular matrix components (e.g. fibronectin and collagens), resulting in fibrosis and disrupting normal tissue architecture, which in the lungs is critical to its primary function, gas exchange.7,8 The pathogenesis of pulmonary fibrosis is complicated and our understanding of it remains inadequate. It involves a number of complex mechanisms, including epithelial/endothelial injury, clotting response, innate and adaptive immunity, fibroblastic cell response, and tissue remodeling. However, full examination of these mechanisms is beyond the scope of this review and is discussed in detail elsewhere.9 –13 Herein, we will focus on the receptor for advanced glycation endproducts (RAGE) and its perplexing role in the pathogenesis of IPF.
RAGE is a member of the immunoglobulin superfamily of receptors and is a pattern-recognition receptor, first discovered for its ability to bind advanced glycation endproducts (AGEs), promoting inflammation and vascular complications in diabetes. 14 RAGE expression is highest in the lung, most notably in alveolar epithelial type I cells (AEC1)15,16 and is a central mediator of inflammation in lung disease. 17 RAGE is highly conserved across mammalian species exhibiting transcriptional variation throughout tissues.18,19 The RAGE protein exists as full-length membrane-bound RAGE (mRAGE), as well as a soluble form (sRAGE), which lacks the transmembrane and signaling domains, and functions as a decoy receptor. sRAGE can be produced endogenously by alternative splicing (esRAGE) or through proteolytic cleavage (cRAGE) by matrix metallopeptidase-9 (MMP9) or a disintegrin and metallopeptidase domain-10 (ADAM10).20 –23 RAGE is a promiscuous receptor, binding various ligands including, high-mobility group box-1 (HMGB1), S100 proteins, β-amyloid peptides and others. 17 RAGE-ligand binding can initiate feed-forward positive feedback, amplifying RAGE-signaling and contributing to the prominent role of RAGE during persistent inflammation.24,25 RAGE-activation induces a multitude of intracellular signaling pathways, contributing to various cellular processes including inflammation, migration and proliferation.26 –29 RAGE has long been linked to IPF; however, puzzling data from both human and experimental studies have left an incomplete and enigmatic story. This perspective review will explore the current understandings of the role of RAGE in IPF, proposing new potential mechanisms of RAGE signaling in fibrosis and how this may relate to RAGE as both a biomarker of disease progression and a potential therapeutic target.
RAGE expression in IPF
It has been well established that overall RAGE expression is significantly decreased in human IPF lungs, as well as mice with experimental pulmonary fibrosis (Figure 1). However, the cause and effect of this phenomenon remains a controversial subject of debate. Initially, Hanford et al. 16 demonstrated that both sRAGE and mRAGE were decreased in the lungs of mice after bleomycin-induced pulmonary fibrosis. Both RAGE protein and messenger ribonucleic acid (mRNA) expression were also decreased in asbestos and crystalline silica exposure models of pulmonary fibrosis;30,31 however, RAGE mRNA levels were not found to be affected in in a subsequent study in mice after bleomycin exposure 32 [Figure 1(b)]. Transcriptomic analyses of human lung tissues indicated that RAGE gene (AGER) transcripts were decreased in subjects with IPF.33,34 AGER expression was lower in the lungs of subjects who presented with more accelerated disease onset (based on time from symptom presentation to diagnosis), while the magnitude of downregulation in AGER transcripts was indistinguishable in both stable IPF and during acute exacerbations.33,34 This suggests that AGER levels may decrease more gradually prior to diagnosis, but remain stably decreased once the disease has progressed enough to be diagnosed. Moreover, multiple studies have shown both mRNA and protein levels of RAGE are significantly reduced in the whole-lung tissue of human subjects with IPF compared with healthy controls30,32 [Figure 1(a)]. Furthermore, in a comparative study of IPF and chronic obstructive pulmonary disease (COPD) lungs, full-length mRAGE and cRAGE were reduced in IPF and COPD lungs, whereas esRAGE was only decreased in IPF lungs. 35 As studies have shown that overall RAGE expression is decreased in IPF lungs, it is important to note that RAGE is most abundantly expressed in AEC1. 36 However, RAGE is expressed at various levels in a number of cell types in the lungs, including airway epithelia, airway smooth muscle cells, lung endothelia, fibroblasts or fibrocytes, alveolar macrophages, and neutrophils.37 –42 Two studies have suggested that RAGE and RAGE ligands were expressed in fibroblastic foci of lungs with UIP.43,44 However, these studies only demonstrated expression of RAGE in fibroblastic foci, and lacked comparison of RAGE expression in UIP compared with normal lung tissues. Another study showed that AGER mRNA levels were decreased in isolated alveolar epithelial type 2 cells (AEC2) cells but not fibroblasts from IPF lungs. 32 Additionally, in vitro stimulation of fibrocytes or fibroblasts with a RAGE ligand enhanced RAGE expression.40,45 Therefore, while data indicate that overall expression of RAGE in the lungs is decreased, this is likely due to loss of AEC1 cells which have the highest level of RAGE expression in the lung, while RAGE expression may persist in lung fibroblasts and fibroblastic foci [Figure 1(a)]. However, too few studies have been done to draw conclusions on the role of RAGE in fibroblastic foci. Therefore, more research is required to clarify the overall expression patterns of RAGE in IPF lungs compared with non-IPF lungs. Studies are needed to establish if there are consistent cell-specific expression patterns of RAGE and if they are associated with disease progression and associated with other biomarker expression patterns in IPF.

RAGE expression in human IPF and in experimental mouse models of pulmonary fibrosis.
s RAGE as a biomarker of IPF disease progression
While analysis of whole-lung homogenates from both humans with IPF and mice with experimental IPF demonstrated a clear decrease in full-length RAGE, sRAGE has also been shown to be decreased.30,32 sRAGE levels were also found to be decreased in the bronchoalveolar lavage fluid (BALF) and plasma of IPF subjects compared with controls46,47 [Figure 1(a)].
A single nucleotide polymorphism (SNP) in the AGER gene, rs2070600, has been associated with natural variation of lung function in people of European ancestry.48,49 This SNP, which results in a glycine–serine amino acid substitution (G82S) in the ligand-binding domain of RAGE results in enhanced RAGE activity.50,51 In addition, rs2070600 has been associated with decreased systemic levels of sRAGE in COPD and severe neutrophilic asthma.52,53 Recently, Manichaikul and colleagues demonstrated that while there was no convincing association of rs2070600 with IPF risk, the minor allele A was associated with lower levels of plasma sRAGE in subjects with IPF and other interstitial lung diseases (ILDs). 54 Moreover, plasma sRAGE levels showed a strong association with disease severity (reduced forced vital capacity precentage), decreased survival, and increased transplantation rates. 54 A similar study also showed an association of decreased plasma sRAGE with the minor T allele, which correlated with acute exacerbation risk and decreased survival rates in IPF. 55 However, it remains unclear if decreased sRAGE levels associated with IPF progression act as a contributing pathological factor, or as a result of the loss of the AEC1 (the major source of sRAGE in the lungs) due to alveolar destruction. Nonetheless, accumulating evidence suggests that circulating sRAGE exhibits strong potential as a prognostic biomarker in IPF. 54
The puzzling role of RAGE in experimental models of IPF
While RAGE signaling is a major component of both homeostatic and pathogenic processes in the lungs, it has also been implicated in the development of fibrosis in the heart, liver, and renal system.56 –58 Although there is mounting evidence that RAGE plays a significant role in pulmonary fibrosis, inconsistent outcomes among different experimental models has been difficult to interpret. Initially, He et al. 59 demonstrated that RAGE-null mice were protected from bleomycin-induced injury and fibrosis, had better survival rates, and lacked production of profibrotic mediators [i.e. transforming growth-factor beta (TGFβ) and platelet-derived growth factor (PDGF)] as compared with wild-type (WT) mice. A later study confirmed these findings and added that mice heterozygous for RAGE also exhibited reduced fibrosis in response to bleomycin 60 [Figure 1(b)]. Conversely, Englert et al. 30 found that RAGE-null mice exhibited exacerbated pathology in response to asbestos-induced injury compared with WT mice, and that RAGE-null mice spontaneously developed fibrotic features with extensive aging [Figure 1(b)]. Alternatively, in response to crystalline silica-induced lung injury, WT and RAGE-null mice exhibited a similar magnitude of fibrosis; however, RAGE-null mice displayed a more diffuse pattern of fibrosis compared with the classic nodular fibrotic pattern seen in WT mice [Figure 1(b)]. In addition, RAGE-null mice exposed to silica had increased lymphocytes and decreased neutrophils and TGFβ levels in the BALF compared with WT mice. 31 Interestingly, intraperitoneal injection of WT mice with lung-derived mouse sRAGE had no effect on bleomycin or asbestos-induced fibrosis. 60 However, this effect may in part be explained by the short half-life of sRAGE in the mouse, where the only effective route of delivery of sRAGE to the lungs was by direct intratracheal instillation. 61 Notably, direct administration of mouse sRAGE intranasally had significant inhibitory effects on house dust-mite-induced asthma in mice. 62 Therefore, it is possible that direct administration of sRAGE or RAGE small-molecule antagonists to the lungs may have inhibitory or therapeutic effects in models of fibrosis, warranting future investigations.
It remains unclear why such differences were seen across different experimental models. However, results from cell and molecular studies offer some insight into the role of RAGE in cellular adhesion/re-epithelialization, and possibly promoting mesenchymal-cell activation. Firstly, as noted earlier, RAGE expression was decreased in AEC2 cells but not fibroblasts from IPF lungs compared with healthy donor lungs. 32 In vitro, genetic inhibition of RAGE had no effects on bleomycin or asbestos-induced cell death, but reduced epithelial cell adhesion, increased proliferation, migration, and wound-healing capacity.32,60 In addition, Zhai et al. 63 recently demonstrated that stimulation of alveolar epithelial cells with AGEs and HMGB1 promoted wound-healing capacity and proliferation in a RAGE-dependent manner. In line with this, previous studies demonstrated that RAGE on AEC1 cells binds to collagen and facilitates cell adhesion/spreading. 64 While there are inconsistencies in the role of RAGE in epithelial-cell adherence, it also promotes epithelial-cell proliferation, migration and wound healing, suggesting a complicated role in re-epithelialization.
Epithelial-to-mesenchymal transition (EMT) has been implicated as a key mechanism in the development of fibrosis, whereby cells lose epithelial characteristics and gain mesenchymal functions to promote fibrogenesis. 65 The role of RAGE in EMT is also controversial, based on mixed findings. For instance, the RAGE ligand HMGB1 induces EMT in mouse AEC2 cells from WT but not RAGE-null mice. 59 However, TGFβ is a potent inducer of EMT and reduces RAGE expression in epithelial cells in vitro.66,67 Conversely, AGE-RAGE signaling has been shown to inhibit TGFβ-induced EMT. 68 Hence, understanding the role of RAGE in EMT remains largely incomplete and likely depends on both initiating and inhibitory factors. Thus far, data suggest that RAGE may be a critical component of re-epithelialization in response to lung injury, while on the other hand, RAGE may also promote processes such as EMT. However, fibroblastic cells are major effector cell types in fibrogenesis, and the role of RAGE signaling in these cells remains poorly understood. The sections below provide new experimental concepts, which, upon investigation, may provide novel insights as to how RAGE signaling promotes fibrosis in the lungs, as well as other tissues.
New prospective mechanisms of RAGE in pulmonary fibrosis
RAGE-mediated regulation of type 2 inflammation
The role of inflammation in the pathogenesis of IPF has long been a subject of fierce debate. Theoretically, it has been thought that unknown insults and environmental exposures cause injury that causes repeated cycles of inflammation and improper repair and remodeling, leading to fibrogenesis. 69 However, lack of robust inflammation in subjects with IPF and ineffectiveness of corticosteroid treatment challenges this theory. 70 Conversely, various studies have demonstrated that inflammatory cell influx is present during exacerbation and is associated with worse disease outcomes.1,71 Moreover, pro-inflammatory and profibrotic cytokines, such as type 2 cytokines interleukin 4 (IL-4) and IL-13 are elevated in subjects with IPF, as are their receptors.72 –74 At mucosal barrier sites, type 2 immunity is a critical defense mechanism against parasitic infections. However, when dysregulated, it can lead to adverse immune responses which drive the development of not only allergic asthma and atopic dermatitis (AD) but also tissue fibrosis.12,75 During such reactions, type 2 cytokines (e.g. IL-4 and IL-13) are produced by various cells including T-helper 2 (Th2) cells, mast cells, and type 2 innate lymphoid cells (ILC2). 76
Of particular interest is IL-13, a pleiotropic type 2 cytokine which promotes inflammation, cell proliferation, and fibrogenesis. 77 In mice, induced over-expression of IL-13 promotes many features of asthma, including subepithelial fibrosis. 78 Transgenic over-expression of IL-13 in the lungs causes subepithelial and adventitial airway fibrosis via induction of TGFβ expression, MMP9-mediated activation of latent TGFβ and collagen deposition.79,80 In vitro, stimulation of pulmonary fibroblasts with rIL-13 signals via signal transducer and activator of transcription 6 (Stat6), inducing target gene expression (e.g. Postn, Col1a1, Pdgfaa, Ccl11, Egr1, Mmp9), TGFβ activation, fibroblast proliferation and collagen production81 –87 [Figure 2(b)]. In addition, IL-13 promotes the alternative polarization of macrophages to a profibrotic ‘M2’ phenotype, which are critical cellular mediators of fibrosis in various tissues. 9 Stimulation of macrophages with IL-13 also signals via Stat6, inducing gene expression of target genes, including Arg1, Relma, Pdgfaa, and Tgfb188,89 [Figure 2(c)]. The critical role of IL-13 in these processes has sparked keen interest in its contribution to lung fibrosis. Indeed, studies demonstrated increased levels of IL-13 in the BALF of subjects with IPF compared with subjects with non-specific interstitial pneumonia or healthy controls.72,90,91 In addition, expression levels of IL-13 receptor subunits are enhanced in fibroblasts from subjects with ILD.72,74 In fibroblasts, IL-13 induces the production of various profibrotic mediators, most notably, periostin, which has been associated with disease progression in IPF90,92,93 [Figure 2(b)]. Periostin is a matricellular protein produced by airway epithelial cells and fibroblasts which binds directly to collagen and cell-surface integrins and promotes collagen fibrillogenesis, cell proliferation, and activation of fibrogenic mediators.94,95 Periostin expression is increased in the lungs of subjects with fibrotic ILD compared with non-fibrotic pneumonias. 93 Moreover, systemic levels of periostin are increased in IPF, show an inverse correlation with lung function indices, and are predictive of pulmonary function decline and survival.93,96 Additionally, mechanistic studies in experimental models suggest that periostin mediates pulmonary fibrosis by promoting airway lung inflammation. 97 In macrophages, IL-13 induces expression of profibrotic mediators such as PDGF, and TGFβ. 98 In addition, IL-13 induces arginase-1, which promotes collagen production through L-arginine metabolism. 88 This is suggestive of local signaling of IL-13 and other profibrotic factors such as periostin, which likely coordinate imbalanced inflammation and tissue repair, and drive pulmonary fibroblast proliferation, activation, and matrix deposition in fibrotic foci [Figure 2(a–c)].
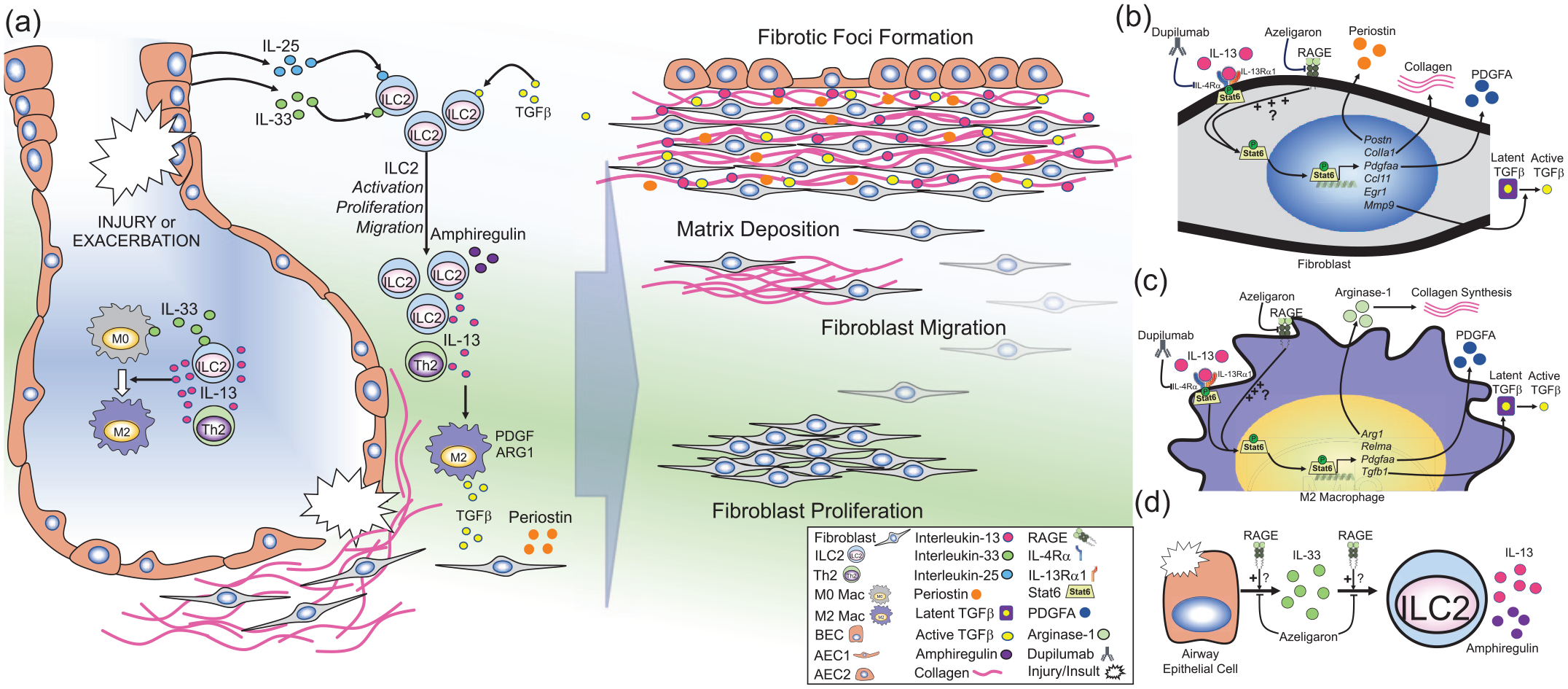
RAGE-mediated regulation of type 2 immunity.
ILC2s have emerged as a unique bridge between the innate and adaptive immune systems, functioning as tissue-resident immune sentinels. 77 Tissue damage and environmental stimuli cause the release of epithelial cytokines (e.g. IL-33, IL-25), which promote the local activation, proliferation, as well as recruitment of ILC2s to the lungs, leading to rapid and coordinated type 2 immune responses. 77 While various cells produce IL-13 (e.g. mast cells and Th2 cells), ILC2s are the most potent source of IL-13 in diseases such as allergic asthma 99 [Figure 2(a)]. Moreover, TGFβ is a critical mediator of ILC2 development, and regulates local ILC2 activity including elaboration of IL-13100,101 [Figure 2(a)]. Recent studies have demonstrated that ILC2s promote pulmonary fibrosis in experimental models of IPF and are increased in subjects with IPF.102 –105 Li and colleagues demonstrated that IL-33-mediated accumulation of ILC2s and M2 macrophages is a driving force in development of fibrosis in response to bleomycin. 104 In addition, IL-33 levels are shown to be increased in the BALF of subjects with IPF compared with healthy controls or other ILD. 106 Moreover, Hams et al. 105 demonstrated that IL-25-mediated activation of ILC2s promotes IL-13-driven lung fibrosis, and that both IL-25 and ILC2s were increased in the BALF of subjects with IPF. Additionally, ILC2s were increased in the circulation of subjects with systemic-sclerosis-related ILD. 103 These studies suggest that similar to asthma and AD, ILC2s are a major producer of IL-13 which is capable of driving pulmonary fibrosis [Figure 2(a)].
The intriguing role of IL-13-mediated processes in the development of pulmonary fibrosis prompted investigation of IL-13 inhibition in a humanized mouse model of IPF in severe combined immunodeficient mice. 90 Murray et al. 90 demonstrated that treatment with an anti-IL-13 antibody (tralokinumab) reduced lung fibrosis and restored markers of epithelialization in mice with established disease. Recent clinical trials have evaluated safety and efficacy of tralokinumab, a bi-specific anti-IL-4/IL-13 antibody (SAR156597), as well as an anti-IL-13Rα1 antibody (lebrikizumab) in IPF; however, none of the trials demonstrated efficacy in primary endpoints.107 –109 A recent experimental follow-up study has suggested that targeting cells expressing both IL-4/IL-13R subunits rather than IL-13 directly may be more effective in reducing fibrotic parameters in the humanized mouse model of IPF. 110 Similarly, in asthma, several clinical trials found that tralokinumab and lebrikizumab failed to meet primary endpoints in the treatment of moderate-to-severe asthma.111 –113 However, recent trials with dupilumab, which targets the shared IL-4/IL-13 receptor subunit (IL-4Rα), has shown strong efficacy in decreasing asthma exacerbations in subjects with severe eosinophilic asthma.114 –116 Dupilumab targets both the type 1 and type 2 IL-4R which are primarily present in immune cells and structural cells, respectively.117,118 IL-4Rα is able to target both IL-4 and IL-13 and block signaling in both structural and immune cells, both of which are key players in asthma and IPF. Therefore, it may be suitable to investigate the effectiveness of dupilumab in models of IPF, with hope that it may be effective in IPF, as seen with eosinophilic asthma (Figure 2).
Recent work from our laboratory and others has demonstrated that RAGE plays a central role in experimental models of type 2 inflammation-driven asthma.37,62,119 –121 RAGE was required at multiple steps in asthma progression, including release of IL-33 in response to allergens, as well as rIL-33 induced accumulation of ILC2s in the lungs 119 [Figure 2(d)]. Moreover, our latest findings revealed that RAGE is also a critical mediator of IL-13-induced signal transduction and consequent airway inflammation. 37 These studies demonstrated that RAGE is required for sustained IL-13-induced Stat6 activation and subsequent upregulation of Stat6-inducible genes (e.g. Clca1 and Ccl11), which drive mucus production and inflammation. 37 In addition, inhibition of RAGE blocks rIL-13 induced expression of periostin in primary human bronchial epithelial cells (unpublished findings). The critical role of RAGE in regulating IL-13 signaling either directly or through ILC2s in models of asthma suggests RAGE may also play a similar role in regulating IL-13-mediated lung fibrosis (Figure 2).
While whole-lung levels of RAGE are decreased in IPF lungs, it is most abundantly expressed in the type I alveolar epithelium, which is decimated in IPF. 122 However, it is possible that RAGE expression is still intact or even enhanced at local sites of active tissue repair or fibrosis. Studies have suggested that RAGE may be expressed in fibroblastic foci in the lungs of subjects with ILD, however more studies are needed to confirm this finding and determine if there is also active signaling.44,46 Moreover, although ILC2s are scarcely distributed in the lungs, local tissue-damage-induced release of epithelial cytokines (e.g. IL-33) causes them to produce copious amounts of IL-13.123,124 Therefore, although alveolar RAGE expression may be lost to injury and lack of adequate re-epithelialization, RAGE signaling may still promote focal inflammation and tissue repair in response to lung injury, leading to fibrotic foci development. Future investigations are needed to evaluate the effects of RAGE inhibition on epithelial initiator cytokines, ILC2s and IL-13 mediated effects in fibroblasts and macrophages in experimental models of pulmonary fibrosis [Figure 2(a–d)].
The calprotectin–RAGE axis in pulmonary fibrosis
In contrast to trends of sRAGE, various RAGE ligands are elevated in lungs and systemically in subjects with IPF, many of which have strong pro-inflammatory effects.44,125,126 The S100 calcium-binding proteins S100A8 and S100A9 are danger signals (or alarmins), which together can form a heterodimer referred to as calprotectin. S100A8/A9 signals through both TLR4 and RAGE and has pro-inflammatory and antimicrobial effects. 127 They are produced mainly by activated inflammatory cells and are elevated in severe neutrophilic asthma and cystic fibrosis (CF) exacerbations.128 –131
S100A8/A9 was shown to be elevated in the BALF of subjects with systemic-sclerosis-related ILD and correlated with the degree of fibrosis. 132 However, a number of studies have demonstrated increased levels of S100A9 in the BALF, but not systemically in subjects with IPF, suggesting it is produced and acts locally in the lungs133 –137 (Figure 3). Enhanced S100A9 levels in IPF correlated with severity of disease, as well as eosinophil and neutrophil numbers133,135,137 (Figure 3). Moreover, elevated S100A9 levels were able to distinguish between IPF and other ILD. 136
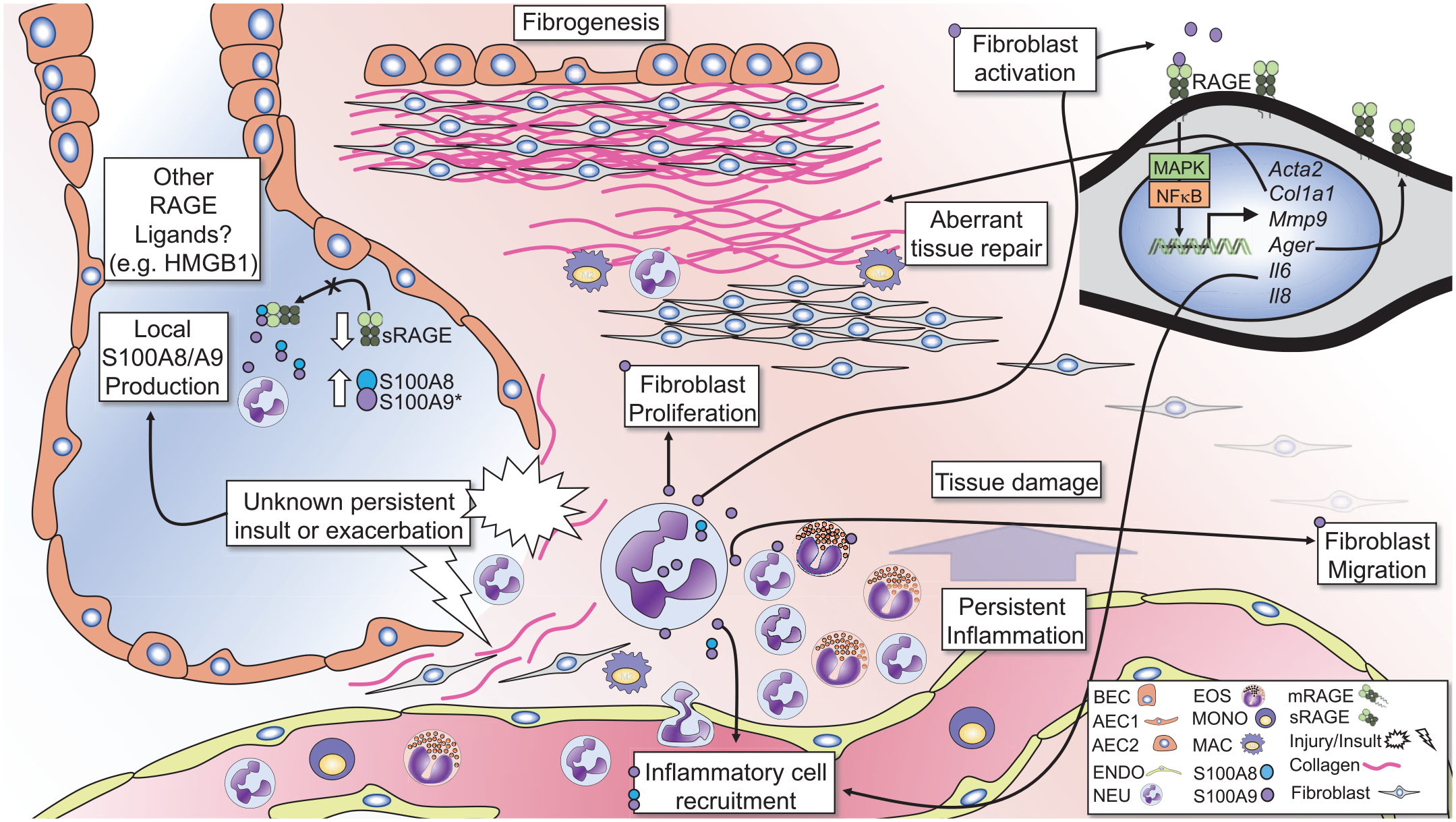
The calprotectin–RAGE axis in pulmonary fibrosis.
A growing body of evidence suggests that local production of S100A8 and S100A9 by inflammatory cells promotes lung inflammation in a feed-forward manner (Figure 3). Recently, a study found that S100A8/A9 drives neutrophil accumulation in the lungs in a mouse model of Mycobacterium tuberculosis infection. 138 Additionally, S100A8/A9 is associated with neutrophils and is a marker of exacerbation in CF, while S100A9 drives neutrophilic inflammation in experimental models of asthma.130,139,140 It is conceivable that S100A8/A9 may contribute to persistent inflammation or exacerbations in IPF, which can promote damage to the lung architecture, initiating progressive lung remodeling (Figure 3).
Experimental studies have demonstrated that S100A9 promotes fibrogenic processes in multiple organs.141 –143 Signaling through RAGE, recombinant (r)S100A9 induced migration and enhanced expression of RAGE, in human fibrocytes 40 and stimulation of fibroblasts with rS100A9-induced proliferation, and increased expression of collagen and α-smooth-muscle actin. 45 rS100A9 also enhanced expression of RAGE and pro-inflammatory cytokines (IL-6 and IL-8) in human lung fibroblasts 45 (Figure 3). Furthermore, S100A8/A9 induces expression of matrix metalloproteinase-9, which, as noted above, cleaves mRAGE to produce sRAGE and activates latent TGFβ, which is a driving force in lung fibrosis. 144
In addition to increased levels of S100A9 in the BALF, immunohistochemical examination of lung sections showed increased S100A9 expression in fibrotic foci. 133 It is also interesting to note that exposure to allergens or direct stimulation with rIL-13 induces production of S100A8/A9 in the lungs of mice.37,145 Active inflammation during stable disease progression or acute exacerbation may promote granulocytic influx, and in turn, further elaboration of inflammatory mediators like S100A8/A9 (Figure 3). This type of feed-forward inflammatory signaling promotes excessive inflammation and lung damage, followed by aberrant repair mechanisms and diminished lung functions in diseases such as pulmonary tuberculosis or CF. These findings suggest that local production S100A8 and/or S100A9 by accumulating immune cells can signal through RAGE on fibroblasts which may contribute to trafficking of fibroblasts to areas of aberrant remodeling, enhancing matrix deposition and fibrogenesis, consequently promoting disease progression (Figure 3).
Nuclear RAGE, cellular senescence, and fibrosis
While RAGE is conventionally known for its function as a pattern-recognition receptor localized at the plasma membrane, recent reports by Kumar et al. have identified a nuclear isoform of RAGE, which has a role in DNA-damage-repair regulation.146,147 Mechanisms that regulate cellular senescence contribute to age-related diseases and progressive fibrosis.148,149 It was shown that in well-aged (~40–50 weeks) RAGE-null mice, fibrotic areas of the lungs had signs of senescence demonstrated by the presence of senescence-associated beta galactosidase.146,147 These recent investigations demonstrated that RAGE can translocate to the nucleus where it is phosphorylated at Ser376 and Ser389 by the ataxia–telangiectasia-mutated kinase. In addition, phosphorylation recruits RAGE to areas of DNA double-strand breaks, where it enhances endonuclease activity, promoting DNA damage repair146,147 (Figure 4). Lack of adequate DNA-damage repair is thought to induce a senescence-associated secretory phenotype (SASP) in fibroblasts, which promotes secretion of pro-inflammatory (e.g. IL-6) and fibrogenic (e.g. TGFβ) mediators, which can drive progression of fibrosis 150 [Figure 4(a)]. It is suggested that lack of RAGE leads to increased cell senescence due to lack of adequate DNA-damage repair, promoting SASP and subsequent lung fibrosis in aged RAGE-null mice 146 (Figure 4). Moreover, data suggest that reconstitution of RAGE may be able to reverse cellular senescence, and in turn, restore lung function, to some degree, in aged mice. 146 Lastly, the authors expanded their studies to show that nuclear RAGE also plays a role in diabetic-associated kidney and lung fibrogenesis. 147 These exciting findings highlight a previously unknown role for RAGE in DNA repair, senescence, and suggest its potential role in regulation these processes may contribute to age-related fibrotic disease. It is noteworthy to mention that bleomycin is used as a chemotherapeutic agent, which induces DNA damage and premature cell senescence. 151 However, RAGE-null mice are completely protected from bleomycin-induced fibrosis.59,60 This presents an interesting paradox, that while both bleomycin-treatment and lack of RAGE expression over time (aging) induce DNA damage, cell senescence and fibrosis, non-aged RAGE-null mice do not develop fibrosis in response to bleomycin-induced injury. Age is a prominent risk factor for IPF in humans. 152 Experimentally, it has been shown that aged mice exhibit impaired resolution of fibrosis compared with younger mice. 153 This reduced ability to resolve fibrosis has been linked to age-related production of senescent and apoptosis-resistant myofibroblasts. 153 In addition, age-related increases in senescent AECs promote fibrosis in aged mice. 154 Likewise, studies have indicated epigenetic regulation of cellular senescence protects younger mice from fibrosis, while promoting fibrosis in aged mice. 155 It is possible that younger RAGE-null mice are resistant to or are able to recover from bleomycin-induced injury in a ‘one hit’ model preventing fibrosis. While on the other hand, continuous injury, or extensive DNA damage from aging without proper DNA-damage repair (due to lack of RAGE), may promote development of fibrosis, over time.

Nuclear RAGE, cellular senescence, and fibrosis.
Studies demonstrating that RAGE-null mice were protected from bleomycin-induced fibrosis were performed in younger mice. To our knowledge, no studies to date have evaluated the expression levels of RAGE in the lungs, over time. It is possible this reduced RAGE expression in aged mice could contribute to the increased susceptibility to, or loss of ability to, resolve fibrosis in response to injury in the lungs over time, leading to cellular senescence and fibrosis. Nonetheless, future research will be needed to determine the role of nuclear RAGE in the pathogenesis of IPF and other fibrotic diseases.
Future implications
It is noteworthy that while recent trials targeting type 2 cytokines in IPF have not yet shown efficacy in improving lung function, such therapies seem more likely to slow progression of disease, by reducing or preventing active fibrogenesis. This notion should be considered in determining the therapeutic potential of other drugs including dupilumab or RAGE antagonists in IPF, which could potentially be used in combination with the currently approved antifibrotics. With RAGE being most abundantly expressed in the alveolar epithelium, of which the primary function is gas exchange, it seems logical that alveolar damage and consequently, reduced RAGE expression, directly correlates with lung function decline in IPF. This suggests that loss of RAGE may be, in effect, a casualty of alveolar injury and loss of normal lung architecture in IPF. On the other hand, developmental studies indicate that RAGE contributes to alveolarization in the lungs, suggesting alternative mechanisms or genetic errors could causally lead to reduced RAGE expression, and in turn, aberrant re-epithelialization.148,149 Nonetheless, evidence suggests that decreased circulating levels of sRAGE have a strong correlation with decline in lung function and may serve as a critical biomarker of disease progression.54,156
The evidence of RAGE ligand signaling in fibroblasts and fibroblastic foci, as well as its role in regulating pro-fibrotic signaling (e.g. IL-13, S100A8/A9) suggest that RAGE may play a causal role in progressive fibrogenesis. RAGE small-molecule inhibitors are shown to be very safe in humans in clinical trials to treat Alzheimer’s disease, suggesting the possible use as a therapeutic. 157 It seems possible that while RAGE neutralization therapies may not necessarily be able to improve lung function, it may be able to inhibit active inflammation and fibrogenesis, thereby halting or slowing disease progression, and prevent acute exacerbations, leading to more long-term and stable disease. Unfortunately, findings from experimental mouse models of pulmonary fibrosis remain puzzling, as genetic ablation of RAGE has both protective and exacerbating effects in these models. Moreover, translation of mouse models to human disease remains a major burden to medical research in general. However, utilization of humanized mouse models of IPF may allow for determination of the capacity of human-safe RAGE inhibitors to improve lung function or slow progression and extend survival in mice with established disease, giving insight to its potential as a human therapeutic for IPF. 110
Interestingly, studies consistently found that genetic ablation of RAGE protects against bleomycin-induced injury in mice.30,59 Bleomycin is a major chemotherapeutic agent, and treatment dosing is limited by the development of pulmonary toxicity and fibrosis. 158 Use of RAGE antagonists may be very useful in enhancing the chemotherapeutic effects of bleomycin by allowing the use of higher doses while preventing bleomycin-induced injury and fibrosis. However, studies are needed to determine the ability of RAGE antagonists to inhibit bleomycin-induced fibrosis or therapeutic effects in established disease.
Lastly, the recent discovery of the function of RAGE in DNA-damage repair and cellular senescence is fascinating, and may suggest that loss of overall RAGE expression contributes to the progression of IPF.146,147 In light of this, it seems plausible that RAGE antagonists (which target the ligand-binding domain) may be able to block the effects of ligand binding without impeding the role of nuclear RAGE, which does not appear to be dependent on the ligand-binding domain. However, further research is required to fully elucidate the mechanisms of RAGE in DNA-damage repair, cell types involved, and how this may contribute to fibrosis in humanized models. Nonetheless, these are exciting findings that may present a new hope in not only halting disease progression, but also the possibility of remission in IPF.
Conclusion
Recent advances in the use of antifibrotics in treatment of IPF have had a significant impact on survival rates. 5 However, incidence and mortality do not wane, and continue to rise, with nearly 50% of patients dying within 5 years of diagnosis, necessitating the need for early detection and novel therapeutic approaches. 159 While the role of RAGE in IPF remains a perplexing and incomplete story, new advances in RAGE biology may provide insight into the confounding findings gathered thus far. Newly illuminated roles of RAGE in alarmin signaling, type 2 immunity, and cellular senescence may link RAGE to mechanisms involved in acute exacerbations and progression of fibrosis in IPF. Moreover, circulating sRAGE levels may present a strong prognostic biomarker and may be linked to genetic susceptibility, which is also present in other chronic lung disorders. While research is needed to fully elucidate the mechanisms by which RAGE contributes to lung injury and fibrogenesis, it remains an intriguing potential biomarker and therapeutic target in IPF.
Supplemental Material
sj-pdf-1-tar-10.1177_17534666211016071 – Supplemental material for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty?
Supplemental material, sj-pdf-1-tar-10.1177_17534666211016071 for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty? by Timothy N. Perkins and Tim D. Oury in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-pdf-2-tar-10.1177_17534666211016071 – Supplemental material for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty?
Supplemental material, sj-pdf-2-tar-10.1177_17534666211016071 for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty? by Timothy N. Perkins and Tim D. Oury in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-pdf-3-tar-10.1177_17534666211016071 – Supplemental material for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty?
Supplemental material, sj-pdf-3-tar-10.1177_17534666211016071 for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty? by Timothy N. Perkins and Tim D. Oury in Therapeutic Advances in Respiratory Disease
Supplemental Material
sj-pdf-4-tar-10.1177_17534666211016071 – Supplemental material for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty?
Supplemental material, sj-pdf-4-tar-10.1177_17534666211016071 for The perplexing role of RAGE in pulmonary fibrosis: causality or casualty? by Timothy N. Perkins and Tim D. Oury in Therapeutic Advances in Respiratory Disease
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: TNP is funded by an American Heart Association Postdoctoral Fellowship award: 19POST34370078. TDO is funded by a Cystic Fibrosis Foundation grant OURY19GO.
Supplemental material
The reviews of this paper are available via the supplemental material section.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.