
Abstract
Cystic fibrosis (CF) is a genetic disorder that causes multiorgan morbidity and premature death, most commonly from pulmonary dysfunction. Mutations in the CF transmembrane conductance regulator (CFTR) gene, of which almost 2000 have been described, result in a dysfunctional CFTR protein. This protein is an adenosine triphosphate binding anion channel, present primarily at the surface of epithelial cells. Loss of function mutations in this anion channel result in decreased or absent chloride/bicarbonate transport. The subsequent abnormal salt and water transport at epithelial cell surfaces leads to thickened secretions, and infection or inflammation in affected organs. In the last 20 years, therapeutics have been developed to treat the signs and symptoms of CF. However, in 2012, the small molecule drug, ivacaftor, became the first approved therapy that addresses the basic defect in CF. Ivacaftor is a potentiator of CFTR channels defective in their chloride/bicarbonate gating/conductance, but present at the epithelial cell surface. It is only approved for 10 mutations carried by approximately 7% of the population of patients with CF. F508del is the most common CFTR mutation, present in homozygosity in approximately 50% of patients with CF. The F508del mutation results in multiple CFTR channel defects that require both correction (stabilization of misfolded CFTR and trafficking to the epithelial cell membrane) and potentiation. This article reviews the in vitro and clinical trial data for the potential use of the potentiator, ivacaftor, and the corrector, lumacaftor, in patients with CF.
Keywords
Background
Cystic fibrosis
Cystic fibrosis (CF) is an autosomal recessive genetic disorder that causes decreased survival with an average age of death of approximately 28 years [CFF, 2013]. CF is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene which encodes the CFTR protein, an adenosine triphosphate binding chloride anion channel that functions at the epithelial cell membrane [Ikuma and Welsh, 2000]. At this point, nearly 2000 CFTR mutations have been described [Toronto, 2015]. To varying degrees, these mutations lead to decreased salt and water transport in mucous membrane lined organs as a result of malfunctioning or absent CFTR channels [Rowe et al. 2005]. In the sinuses and lungs, this decreased salt and water transport results in thickened mucus that impairs the body’s ability to clear secretions [Gibson et al. 2003]. A cycle of chronic infection and inflammation leads to tissue destruction [Gibson et al. 2003]. In the gastrointestinal tract, this abnormal mucosal surface fluid regulation leads to exocrine pancreatic dysfunction, malabsorption and intestinal obstruction [Borowitz and Gelfond, 2013].
Modulator therapy
When the CFTR gene was discovered in 1989 [Kerem et al. 1989; Riordan et al. 1989; Rommens et al. 1989], the promise of a cure with gene therapy seemed imminent. However, available gene transport vectors proved inefficient, and clinical trials were not successful [Griesenbach and Alton, 2013]. Subsequently, CF-specific therapeutics that treat the signs and symptoms of CF lung disease were developed, including dornase alfa [Fuchs et al. 1994] (to decrease the viscosity of sputum), and inhaled tobramycin [Ramsey et al. 1999] and inhaled aztreonam [Oermann et al. 2010] (anti-pseudomonal antibiotics). Additional therapies that were tested in clinical trials that treat infection and inflammation [Konstan et al. 1995; Saiman et al. 2003, 2010], and improve mucus clearance [Donaldson et al. 2006; Elkins et al. 2006] have been incorporated into the daily management of this chronic disease. In combination with nutritional support, and comprehensive, coordinated care in accredited CF centers, these therapies improved the survival of patients with CF [Konstan et al. 2010; CFF, 2015]. However, it was not until recently that CFTR modulators (Figure 1), drugs that directly address the basic defect in CF, were tested and proven in clinical trials.

Small molecule modulators of cystic fibrosis transmembrane conductance regulator (CFTR) function include potentiators and correctors. For some mutations, combination therapy will be necessary. Adapted from CFF [2015].

Based on phase III data, absolute improvement in FEV1 % predicted in patients receiving ivacaftor (blue bars) or lumacaftor–ivacaftor combination (rust bar) with one or more of the following CFTR mutations: G551D [Ramsey et al. 2011], non-G551D gating mutation [De Boeck et al. 2014], R117H [NIH, 2015] (Trial Identifier for NIH 2015: NCT01614457) or F508del homozygotes [Wainwright et al. 2015]. #Subjects at least 18 years of age; *pooled data from TRAFFIC and TRAFFIC for subjects receiving lumacaftor 400 mg every 12 h and ivacaftor [Wainwright et al. 2015]. CFTR, cystic fibrosis transmembrane conductance regulator; FEV1, forced expiratory volume in 1 s.
Ivacaftor, a small molecule discovered through high throughput screening, is a CFTR potentiator (Figure 1): it increases the open probability of CFTR channels present at the epithelial cell surface that are defective in their gating, such as the G551D mutation [Van Goor et al. 2009]. Ivacaftor was tested in a phase II multidose, multicenter placebo-controlled randomized trial in 39 adults with CF [Accurso et al. 2010]. Ivacaftor was shown to improve chloride transport both by nasal potential difference measurement (NPD) and sweat chloride concentration, with a median decrease in the level of sweat chloride of −59.5 mmol/liter (range −66.0 to −19.0; p = 0.008 within subject, p = 0.02 versus placebo) at 28 days. A median increase in forced expiratory volume in 1 s (FEV1) of 8.7% (range 2.3–31.3; p = 0.008 for the within-subject comparison, p = 0.56 versus placebo) at day 28 was also achieved in the ivacaftor-treated subjects (Table 1). For the first time since CF was described [Andersen, 1938], these data provided proof of concept that treatment of the molecular defect was possible.
Summary of absolute changes compared to placebo in clinical outcomes based on modulator therapy and mutations targeted.
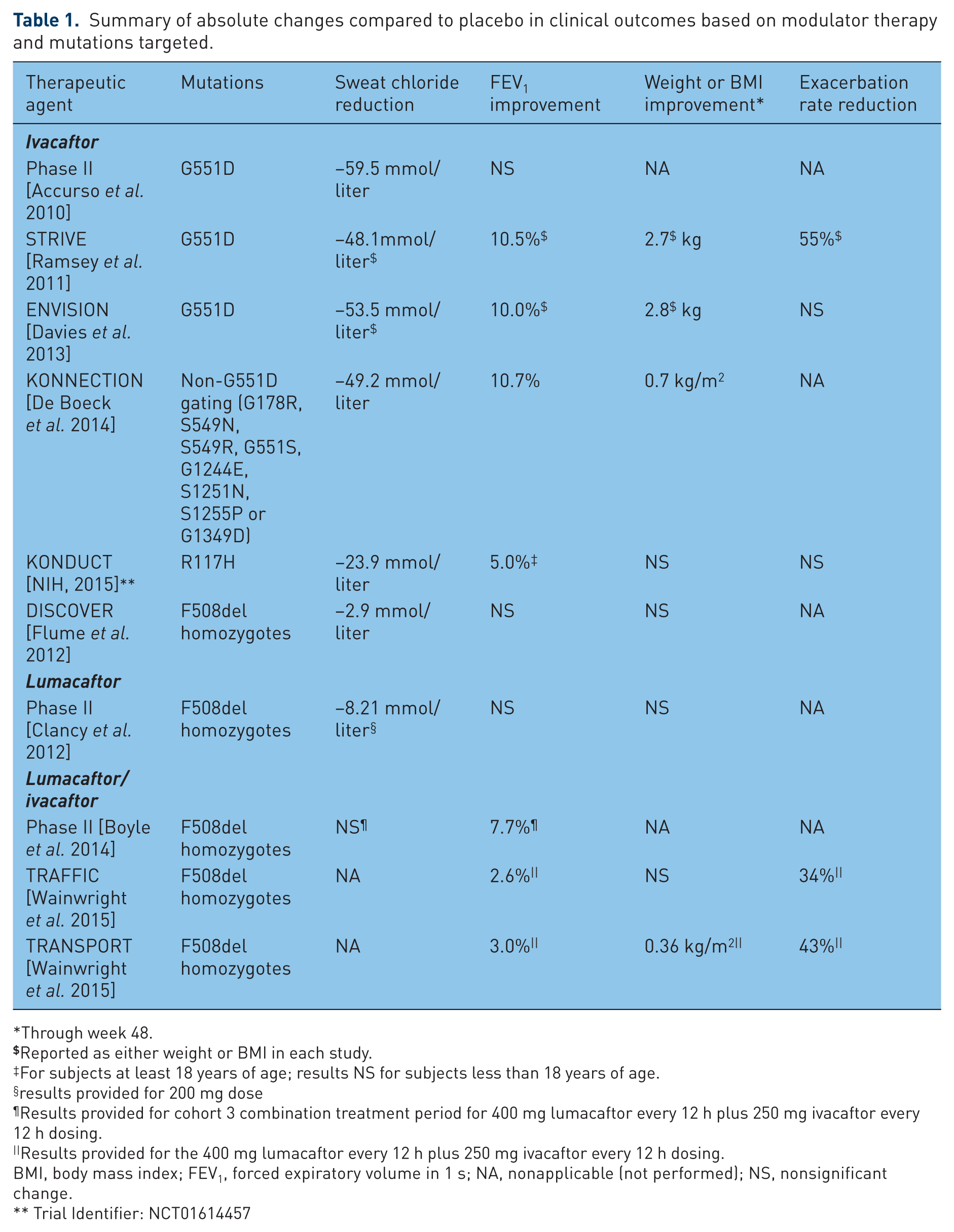
Through week 48.
Reported as either weight or BMI in each study.
For subjects at least 18 years of age; results NS for subjects less than 18 years of age.
results provided for 200 mg dose
Results provided for cohort 3 combination treatment period for 400 mg lumacaftor every 12 h plus 250 mg ivacaftor every 12 h dosing.
Results provided for the 400 mg lumacaftor every 12 h plus 250 mg ivacaftor every 12 h dosing.
BMI, body mass index; FEV1, forced expiratory volume in 1 s; NA, nonapplicable (not performed); NS, nonsignificant change.
Trial Identifier: NCT01614457
Two large phase III studies were conducted in subjects with CF and at least one copy of the G551D mutation: one in subjects at least 12 years of age with FEV1 % predicted of at least 40% and up to 90% (STRIVE) [Ramsey et al. 2011], and one in subjects at least 6 years of age with FEV1 % predicted of at least 40% up to 105% (ENVISION) [Davies et al. 2013]. Data from these studies confirmed the results seen in the phase II study: rapid improvement in sweat chloride and lung function in the first 2 weeks of the study, as well as in weight, quality of life and rate of exacerbations that were sustained over the 24 weeks of the study (Table 1). The open-label roll-over study (PERSIST) [McKone et al. 2014] showed that results from STRIVE and ENVISON were sustained over 96 weeks. Based on the data from the phase III studies, ivacaftor was approved in 2012 for patients with the G551D mutation. This approval marked ivacaftor as the first drug in the history of CF therapeutics to be approved for treatment of the molecular defect in CF. Utilizing data from expanded access programs, ivacaftor was subsequently shown to be beneficial and well tolerated, even in those with advanced disease [Hebestreit et al. 2013; Barry et al. 2014; Taylor-Cousar et al. 2015]. Furthermore, GOAL investigators prospectively collected data on patients who were eligible to receive ivacaftor following US Food and Drug Administration (FDA) approval. In their longitudinal cohort study, the GOAL investigators showed that significant improvements in lung function, weight, and hospitalizations were achievable in patients with the G551D mutation who take ivacaftor even outside of the research setting [Rowe et al. 2014].
The G551D mutation is the most common gating defect (occurring in approximately 4% of the CF population), but there are other, more rare mutations (occurring at a combined rate of approximately 1%) that are also categorized in this class [Boyle and De Boeck, 2013]. In vitro data suggested that ivacaftor could also be efficacious for patients with other gating mutations [Yu et al. 2012]. Ivacaftor was tested in an international multicenter two-part clinical trial (KONNECTION) for patients with one of eight other gating defects (G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P or G1349D) [De Boeck et al. 2014]. Subjects were initially randomized in a blinded fashion to one of two sequences (beginning with ivacaftor or placebo) for 8 weeks, followed by a 4–8-week washout period prior to crossing over to the opposite sequence. All subjects were then given open-label ivacaftor in a 16-week extension period. Subjects experienced a 7.5% improvement in absolute change in FEV1 % predicted (p < 0.0001) at 8 weeks, as well as in sweat chloride, body mass index (BMI), and quality of life. These results were sustained over the course of the extension period. Finally, based on in vitro work suggesting potential response of the conductance defect causing (‘residual function’), mutation R117H [Van Goor et al. 2012], an additional phase III randomized, double-blinded 24-week clinical trial [ClinicalTrials.gov identifier: NCT01614457] using ivacaftor was conducted in children and adults with at least one copy of the R117H mutation. Sixty-nine patients were randomized. Although the study did not meet its primary endpoint (absolute change from baseline in FEV1 % predicted) in the group as a whole, prespecified subgroup analysis in those subjects at least 18 years of age (n = 46) showed an absolute mean improvement in FEV1 % predicted of 5.0 (p = 0.01). Statistically significant improvements were seen in secondary endpoints, including sweat chloride and patient-reported respiratory symptoms. Based on these results, ivacaftor was approved for use in December 2014 in patients with CF who are at least 6 years of age with the R117H mutation.
Ivacaftor is clearly efficacious in patients with CF who have gating mutations or the conducting mutation R117H; however, this indication affects only 6.8% of the US CF population [CFF, 2013]. Approximately 90% of patients with CF are heterozygous for the F508del mutation and nearly 50% are homozygous for this mutation [CFF, 2013]. Compared with patients who were homozygous for the F508del mutation, those who were heterozygous for the mutation and those without the F508del mutation had lower adjusted risk for death [14%, 95% confidence interval (CI) 7–20%; and 25%, 95% CI 15–34%, respectively] [Mackenzie et al. 2014]. Unlike gating or conductance mutations, the F508del mutation causes protein misfolding that results in degradation in the endoplasmic reticulum [Thomas et al. 1992]. Thus, only a small amount of CFTR protein is trafficked to the cell surface [Denning et al. 1992]. Additionally, once at the cell surface, the small amount of F508del CFTR that is present demonstrates defective gating and increased turnover.
In vitro work in F508del homozygous human bronchial epithelial human epithelial cells (HBEC) showed that ivacaftor improved CFTR-mediated chloride secretion to approximately 10% of non-CF HBEC (compared with an increase in F508del/G551D HBEC from 5% to approximately 50% of that observed in non-CF HBEC) [Van Goor et al. 2009]. At that time it was unclear how closely in vitro response would correlate with clinical response. Thus, ivacaftor monotherapy was attempted in a 16-week multicenter, randomized, double-blind, placebo-controlled trial (DISCOVER) of 140 patients homozygous for the F508del mutation [Flume et al. 2012]. As expected, based on the multiple defects that must be corrected to render the F508del protein functional (folding, processing, conductance), the randomized controlled trial of ivacaftor monotherapy did not show improvements in lung function or other clinical endpoints (although there was a clinically insignificant but statistically significant improvement of −2.9 mmol/liter in sweat chloride; 95% CI −5.6 to −0.2, p = 0.0384). Based on these results, use of ivacaftor monotherapy in patients with CF who are homozygous for the F508del mutation is specifically excluded in the ivacaftor package insert [Vertex Pharmaceuticals, 2015].
Solely using the CFTR potentiator, ivacaftor, was insufficient to treat patients homozygous for the F508del mutation. However, data in HBEC looked promising using a CFTR corrector [Van Goor et al. 2011], a drug that improves processing and trafficking of defective CFTR to the epithelial cell surface (Figure 1). Following high throughput screening of 164,000 small molecules for their ability to improve the amount of functional F508del CFTR at the cell surface, Van Goor and colleagues tested the most efficacious compound, VX-809, in cultured HBEC from patients with CF homozygous for the F508del mutation [Van Goor et al. 2011]. They demonstrated that lumacaftor (VX-809) increased processing of F508del CFTR such that an increased amount of F508del CFTR was able to leave the Endoplasmic reticulum (ER) and be trafficked to the cell surface; chloride secretion was improved to approximately 14% of that of non-CF HBEC. Subsequently, these investigators treated the HBEC with both lumacaftor and ivacaftor. The combination increased F508del CFTR chloride secretion to approximately 25% of that of non-CF HBEC. These results suggested that using the corrector to improve processing and trafficking of F508del CFTR to the cell surface, in combination with the potentiator to increase the conductance of F508del CFTR at the cell surface, would potentially lead to a therapy that could affect at least 50% of the CF population.
Pharmacokinetics
While the combination of lumacaftor and ivacaftor in vitro leads to increased F508del CFTR chloride secretion acutely, subsequent in vitro studies showed that prolonged (24–48 h) administration of the two drugs resulted in a drug interaction that reduced the efficacy of lumacaftor. Specifically, in HBEC homozygous for the F508del CFTR mutation, coadministration of ivacaftor with lumacaftor led to decreased folding efficiency and stability of F508del CFTR, resulting in an increased turnover rate and reduction in mature CFTR at the cell surface [Veit et al. 2014; Cholon et al. 2014].
Cohort 1 in the phase II trial was designed to assess the potential pharmacokinetic (PK) interactions between the two drugs in order to aid with dosing parameters, and cohorts 2–3 were conducted to determine whether higher doses and longer duration of treatment might provide clinical benefit that would support phase III trials in spite of the drug–drug interaction [Boyle et al. 2014]. PK findings from the study were submitted to the FDA and were reported by the FDA Clinical Pharmacology Reviewer during the Pulmonary-Allergy Drugs Advisory Committee (PADAC) meeting [FDA, 2015]. Results from the comprehensive clinical pharmacology program included the PK and metabolism of the individual drug components lumacaftor and ivacaftor as well as the combination products.
Steady-state plasma concentrations of the combination of lumacaftor and ivacaftor following twice daily dosing were typically reached after approximately 7 days of treatment [FDA, 2015]. The lumacaftor accumulation ratio was approximately 1.9, whereas that of ivacaftor was lower at day 1 due to the cytochrome P450 3A (CYP3A) induction effect of lumacaftor. Drug absorption of the combination product was also affected by the intake of fatty foods with lumacaftor exposure being approximately two times higher and ivacaftor exposure being three times higher when compared with a fasting state.
Both lumacaftor and ivacaftor are 99% bound to plasma proteins. Lumacaftor is not extensively metabolized so that most of the drug is excreted unchanged in the feces. It is a strong inducer of CYP3A enzymes and has a terminal half life of approximately 26 h. In contrast to lumacaftor, ivacaftor is primarily metabolized by CYP3A enzymes and excreted in the feces with a terminal half life of approximately 12 h. Considering that lumacaftor is a strong inducer of CYP3A, and ivacaftor is a CYP3A substrate, when dosed together ivacaftor exposure is reduced by lumacaftor in a dose-dependent manner. Lumacaftor exposure is not affected by ivacaftor. Findings of this clinically relevant drug–drug interaction were noted in the phase II PK analysis that found when lumacaftor 400 mg every 12 h was administered with ivacaftor 150 mg twice daily, exposure is reduced by more than 80% [Clancy et al. 2012; FDA, 2015].
In vitro studies have also suggested that lumacaftor has the potential to induce CYP2B6, CYP2C8, CYP2C9, and CYP2C19 as well as inhibit CYP2C8 and CYP2C9 [FDA, 2015]. Because of the induction/substrate effects of the combination of lumacaftor and ivacaftor, the metabolism of other drugs that are metabolized by these enzymes may be altered. Therefore, many medications utilized in the management of patients with CF such as oral and intravenous antibiotics, antifungals, proton pump inhibitors, and antidepressants will require dose adjustment if used when a patient is on combination therapy [FDA, 2015].
Current evidence
Phase IIa trial
Based on the promising in vitro evidence of improvement of the function of F508del CFTR following combination treatment of F508del HBEC, investigators designed a phase IIa multicenter, randomized, placebo-controlled trial in adult patients with CF who were homozygous for the F508del mutation to evaluate the safety, tolerability, and PK of lumacaftor (VX-809) [Clancy et al. 2012]. Secondary outcome measures aimed at evaluation of the effect of lumacaftor on CFTR function included sweat chloride and NPD. Lung function and quality of life were also measured as secondary outcomes. Subjects were enrolled into two cohorts. In cohort A, 15 subjects were randomized in a 2:2:1 ratio to receive lumacaftor at 25 mg or 50 mg, or placebo for 28 days. Following safety review of data from the first 15 subjects by an independent data monitoring committee, 74 subjects were randomized in a 2:2:1 ratio to receive lumacaftor at a dose of 100 mg or 200 mg, or placebo for 28 days.
Treatment with lumacaftor led to a dose-dependent decrease in sweat chloride values (p = 0.0013) that occurred within 7 days of drug dosing, and was sustained over the course of the trial. The mean change from baseline in sweat chloride in the highest dose (200 mg) group was −6.6 mmol/liter (95% CI −10.27 to −2.83, p = 0.0008)at 7 days, and −8.21 mmol/liter (95% CI −14.33 to −2.10) at 28 days. This difference was statistically different versus placebo (p < 0.01). Sweat chloride levels returned approximately to baseline levels following a 7-day drug washout period. Although improvements were seen in sweat chloride levels, there were no differences in CFTR-dependent NPD measurement in any of the dose groups. In addition, there were no changes in relative amounts of mature versus immature forms of F508del CFTR in the subgroup of patients (n = 34) who underwent rectal biopsy. Finally, there were no differences in lung function or quality of life in any of the dose groups, although the study was not powered to detect such differences.
Generally, treatment with lumacaftor (VX-809) was safe and well tolerated. The most commonly reported adverse events in lumacaftor- and placebo-treated subjects were cough, headache, and dyspnea. Cough and dyspnea occurred in placebo- versus lumacaftor-treated subjects at rates of 41% and 5.9%, and 46% and 19%, respectively. No subjects in the placebo group withdrew from the study, although one subject in each of the lumacaftor dose groups withdrew from the study. The rate of adverse respiratory events was similar between the different dose groups. Finally, there was no statistically significant difference between rate of exacerbations between the placebo-treated and lumacaftor-treated subjects (12% versus 17%, p = 0.62).
Data from this phase IIa trial showed that modulation of CFTR function in patients homozygous for the F508del CFTR mutation was safe and possible, but insufficient to improve lung function over 28 days. The question remained whether administration of a combination of the corrector and the potentiator could further improve CFTR function sufficiently to improve clinical outcomes.
Phase II trial
Based on the results of the phase IIa trial, researchers designed a multicenter, multidose, randomized, placebo-controlled trial of ivacaftor plus lumacaftor (VX-809) in adults with CF homozygous and heterozygous for the F508del mutation [Boyle et al. 2014]. Three successive cohorts were planned, with dosing for each subsequent cohort defined by data from the previous cohort (Figure 3). The primary outcomes for the study were change in sweat chloride concentration during combination therapy, and safety. Secondary outcomes included change in lung function and change in sweat chloride from baseline in all cohorts. Change in Cystic Fibrosis Questionnaire-Revised (CFQ-R) scores was included as a secondary endpoint in cohorts 2 and 3.

Randomization and dosing schema for cohorts 1–3 in the phase II trial of treatment of subjects homozygous for the F508del mutation with lumacaftor and ivacaftor. Adapted from Boyle et al. [2014].
In cohort 1 (n = 62 subjects randomized) subjects’ FEV1 did not change significantly compared with placebo in either of the lumacaftor plus ivacaftor dose groups. However, during the combination therapy treatment period, cohort 1 subjects who received 200 mg lumacaftor daily and 250 mg lumacaftor twice daily experienced a decrease in sweat chloride of −9.1 mmol/liter (95% CI −12.9 to 5.4, p < 0.001) that was significantly different from that in the placebo group (−9.7 mmol/liter, 95% CI −14.8 to −4.6, p < 0.001). Based on these data, subjects in cohorts 2 and 3 randomized to the study drug were given a combination of lumacaftor and ivacaftor 250 mg every 12 h.
Subjects in cohort 2 (n = 109 randomized) who were homozygous for the F508del CFTR mutation (n = 82) and received either 600 mg daily or 400 mg twice daily of lumacaftor plus 250 mg ivacaftor every 12 h experienced a mean increase in absolute FEV1 % predicted (6.2, 95% CI 3.3–9.0, p < 0.001; and 6.1, 95% CI 2.0–10.2, p = 0.0040, respectively) during combination treatment (but not during monotherapy). These changes in FEV1 were significantly improved compared with placebo in these treatment groups (p < 0.001 and p =0.003, respectively). Mean sweat chloride concentrations did not decrease significantly in any of the groups during the combination therapy period, although there were significant decreases over the entire study period (days 1–56) in the patients homozygous for the F508del CFTR mutation in the daily lumacaftor plus ivacaftor groups (−9.1 mmol/liter, 95% CI −13.3 to −4.9, p < 0.001; and −8.9 mmol/liter, 95% CI −13.1 to −4.7, p < 0.001, respectively). Over the total treatment period, improvement in the respiratory portion of the CFQ-R was observed compared with the placebo group in the subjects who received lumacaftor 400 mg daily (13.5, 95% CI 3.2–23.9, p = 0.011), but not in those who received 600 mg daily. There were no statistically significant differences in sweat chloride or in lung function during combination therapy for subjects heterozygous for the F508del CFTR mutation.
For subjects in cohort 3 (n = 15 patients randomized) homozygous for the F508del CFTR mutation randomized to who received twice daily lumacaftor plus twice daily ivacaftor group,mean sweat chloride concentrations did not decrease significantly during the combination therapy period, although there was a significant decrease in sweat chloride levels over the entire study period (days 1–56) (–10.3, 16.7–4.0, p = 0.002). There was an improvement in lung function during the combination period for subjects in this cohort: a mean absolute increase in FEV1 % predicted of 6.1 (95% CI 2.0–10.2, p = 0.004), and a mean relative increase (compared with placebo) of 7.7 (95% CI 2.7–12.6, p = 0.003). There was not a statistically significant improvement in CFQ-R respiratory scores for subjects in this cohort over the 56-day study period compared with placebo.
Although the incidence of adverse events was similar in the combination and placebo groups, there was a dose-dependent trend towards a decline in FEV1 % predicted in the 100 mg and 200 mg lumacaftor monotherapy treatment groups compared with placebo. Seven subjects receiving lumacaftor monotherapy discontinued treatment because of adverse events, mostly respiratory in nature (dyspnea, categorized as ‘respiration abnormal’, and chest tightness). The most common adverse events during combination therapy treatment were cough, pulmonary exacerbation, and headache. The new onset of chest tightness and dyspnea were not more common than in placebo in subjects on combination therapy and no subjects discontinued treatment because of an adverse event during combination therapy. The rate of serious adverse events, including pulmonary exacerbation, was similar between the combination therapy and placebo groups. There were no serious adverse events corresponding to liver function test abnormalities that were caused by study drug treatment.
The phase II trial revealed a number of important points for consideration in design of the phase III trials. Critically, although the improvements in CFTR function and lung function were modest compared with those observed in the studies of ivacaftor in patients with gating and conductance mutations, the phase II trial showed that modulation of F508del CFTR is possible. Second, this study provided information about dosing, specifically that the 600 mg daily or 400 mg twice daily doses of lumacaftor were most likely to be efficacious. Additionally, in spite of the drug–drug interaction between lumacaftor and ivacaftor [Cholon et al. 2014; Veit et al. 2014], clinical improvements were observed during combination therapy [Boyle et al. 2014]. Although increased respiratory adverse events were observed with higher doses of lumacaftor in some subjects during monotherapy, combination therapy was overall safe and well tolerated. Finally, although the trends for patients heterozygous for the F508del mutation were similar to those homozygous for the mutation, the data demonstrated that this combination of modulator therapies was not sufficiently efficacious in heterozygotes to move forward with testing in this group.
Phase III trial
Using the results of the phase II trial to guide them, investigators designed two phase III clinical trials of lumacaftor–ivacaftor combination therapy in patients at least 12 years of age who were homozygous for the F508del mutation [Wainwright et al. 2015]. The two studies, TRAFFIC and TRANSPORT, were run in parallel. Subjects from 187 multinational sites were randomized in a 1:1:1 fashion to one of the two most promising doses of lumacaftor, 600 mg once daily or 400 mg every 12 h, in combination with ivacaftor 250 mg every 12 h, or placebo for 24 weeks. Randomization was stratified by age, sex, and pulmonary function at screening. The primary endpoint for both studies was the absolute change from baseline in the FEV1 % predicted at 24 weeks. Secondary endpoints for both studies included the relative change from baseline in the FEV1 % predicted, absolute change from baseline at week 24 in BMI and CFQ-R respiratory domain, percentage of patients with a relative increase of at least 5% in FEV1 % predicted, and number of pulmonary exacerbations over the 24 weeks of the study.
Of the 1122 patients who were randomized, 1108 received at least one dose of study drug. The mean age of the subjects across the dosing groups was approximately 25 years of age. Approximately half of the subjects were women, and their baseline FEV1 % predicted was around 61%. Consistent with this degree of lung dysfunction, the majority of patients were concomitantly treated with bronchodilators, dornase alfa, inhaled antibiotics, azithromycin inhaled hypertonic saline, and inhaled glucocorticoids.
In this heavily treated patient population, combination therapy led to modest improvements in the primary and secondary endpoints of the study. Compared with placebo, the mean absolute change in FEV1 % predicted from baseline to week 24 was significantly improved in all dose groups in the individual studies (TRAFFIC: 2.6–4%, TRANSPORT: 2.6–3%, p < 0.001), and in the pooled data (2.8–3.3%, p < 0.001). The relative change in FEV1 % predicted from baseline to 24 weeks was also significantly improved in all dose groups in both studies (TRAFFIC: 4.3–6.7%, TRANSPORT: 4.5–5.3%, p < 0.001) and in the pooled data (4.8–5.6%, p < 0.001). These changes in FEV1 occurred within 15 days of the start of the study drug and were sustained through the 24 weeks of the trial. Furthermore, the improvement in FEV1 % predicted was consistent across all subgroups tested, including age (children versus adults ⩾ 18 years), severity of lung disease (mild, moderate, or severe), and sex and Pseudomonas aeruginosa infection status. Finally, based on the pooled data, the odds ratio for a relative increase of at least 5% from baseline in FEV1 % predicted was 2.2 (95% CI 1.6–3.1, p < 0.001) for the subjects receiving 400 mg twice daily plus ivacaftor, and 3.1 (95% CI 0.8–5.3, p < 0.001) compared with placebo.
In addition to modest improvements in lung function, there were also improvements in important secondary endpoints. Most remarkably, compared with placebo, there was a 30% lower rate of protocol-defined pulmonary exacerbations in the 600 mg lumacaftor group (p = 0.001) and a 39% lower exacerbation rate in the 400 mg twice daily lumacaftor–ivacaftor group (p < 0.001). Notably, compared with placebo, there was a higher proportion of subjects who were exacerbation free at the end of the 24-week trial in the pooled lumacaftor–ivacaftor dosing groups. Pulmonary exacerbations leading to intravenous antibiotics or hospitalization were also reduced in the lumacaftor–ivacaftor treated groups compared with placebo [lumacaftor 600 mg daily plus ivacaftor: 45% reduction in events leading to intravenous antibiotic therapy (p < 0.001) and 39% reduction in events leading to hospitalization (p = 0.003); lumacaftor 400 mg every 12 h plus ivacaftor: 56% reduction in events leading to intravenous antibiotic therapy (p < 0.001) and 61% reduction in events leading to intravenous antibiotic therapy (p < 0.001)]. Also based on pooled data from the two studies, there were statistically significant improvements in absolute change from baseline in BMI in both dosing groups (0.24–0.28, p < 0.001). Finally, although there was a statistically significant 2.2–3.1-point improvement in the respiratory portion of the CFQ-R (p = 0.05 and 0.007 for the lumacaftor 400 mg ivacaftor group and 600 mg ivacaftor group, respectively), this improvement does not meet the previously defined minimally important clinical difference for this outcome [Quittner et al. 2009].
Safety and tolerability results were consistent with those seen in the phase II studies. In general, similar rates of adverse events (most commonly pulmonary exacerbations, cough, and headache) were observed in the placebo and lumacaftor–ivacaftor groups. However, dyspnea and chest tightness were more often seen in those treated with lumacaftor–ivacaftor (13.0–14.9% and 8.7–10.8%, respectively) than in placebo-treated subjects (7.8% and 5.9%, respectively). Furthermore, the proportion of subjects who discontinued treatment was higher in those receiving lumacaftor–ivacaftor. It is noteworthy that for those subjects that chose to continue in the study in spite of the early emergence of respiratory-related adverse events, the respiratory symptoms generally resolved within 2–3 weeks of continued treatment. The most common serious adverse event in all treatment groups was pulmonary exacerbation, which occurred more frequently in the placebo group (24.1% of the patients versus 13.0% in the pooled lumacaftor–ivacaftor group). Rates of clinically significant elevations of liver function tests were similar in the placebo and lumacaftor–ivacaftor treated subjects (5.1% versus 5.2%). However, while there were no liver-related serious adverse events in the placebo group, seven events occurred in the subjects receiving combination therapy. Liver function returned to baseline in six of seven patients following discontinuation or interruption of lumacaftor–ivacaftor therapy.
In summary, based on the data from two phase III studies, lumacaftor–ivacaftor combination therapy was safe and relatively well tolerated with the exception of a small percentage of patients who experienced increased dyspnea and chest tightness. Combination therapy modestly improved lung function and led to clinically important decreased rates of pulmonary exacerbations in subjects homozygous for the F508del mutation. Outcome data were similar for the two lumacaftor–ivacaftor dosing groups with the exception that the effect on exacerbations was stronger for the lumacaftor 400 mg plus ivacaftor group. Based on these data, in November 2014, Vertex Pharmaceuticals submitted a New Drug Application (NDA) for approval of the combination of lumacaftor–ivacaftor (lumacaftor 400 mg and ivacaftor 250 mg every 12 h) in patients with CF who are at least 12 years of age and are homozygous for the F508del mutation.
Future directions
On 12 May 2015, the PADAC to the FDA held a meeting to review safety, efficacy, and the potentially appropriate use of lumacaftor–ivacaftor therapy. Physicians with expertise in CF and patients who had participated in the clinical trials spoke at the hearing. The PADAC voted 12–1 to approve the lumacaftor–ivacaftor combination (proposed trade name, Orkambi) for use in people with CF who are at least 12 years of age and are homozygous for the F508del mutation. Under the Prescription Drug User Fee Act, the FDA approved the Orkambi (Vertex Pharmaceuticals Boston, MA) NDA 206038 on 2 July [FDA, 2015].
Although only very limited data from trials of ivacaftor in patients with FEV1 less than 40% were available at the time of its FDA review, ivacaftor was approved for patients with the G551D mutation without a lung function restriction [Vertex Pharmaceuticals, 2015]. As in the case with ivacaftor, and most studies of new therapeutic agents in patients with CF, patients with severe disease were excluded from the phase III studies of lumacaftor–ivacaftor [Wainwright et al. 2015]. Because treatment options for this group of patients are limited, patients with severe CF who are homozygous for the F508del mutation will be very interested in trying this new therapy. Particularly in light of the increased incidence of chest tightness in patients in the treated versus placebo group [Wainwright et al. 2015], safety and tolerance data in the group of patients with severe CF lung disease are crucial. A 6-month open-label study of lumacaftor–ivacaftor therapy in subjects homozygous for the F508del mutation who are over 12 years with FEV1 less than 40% or actively listed for lung transplant is currently enrolling [ClinicalTrials.gov identifier: NCT02390219].
Lack of validated surrogate endpoints in children under the age of 6 years makes it difficult to prove efficacy in this group. However, safety was shown in a clinical trial of ivacaftor in children aged 2–5 years old with gating mutations [ClinicalTrials.gov identifier: NCT01705145]. This trial led to approval of ivacaftor for patients at least 2 years old with CF with gating mutations [Vertex Pharmaceuticals, 2015]. Because it is clear that lung disease is present from a very young age [Stick et al. 2009], it will be important to conduct safety and efficacy studies in 6–11-year-old patients and safety studies in patients under the age of 5 years.
With its approval, lumacaftor–ivacaftor combination therapy is the first modulator therapy approved that will potentially treat almost half of the patients with CF. Though the effects on important clinical endpoints including lung function and BMI were small [Wainwright et al. 2015], it is a first step. Given the complex pathophysiology of F508del CFTR mutation dysfunction, two or more small molecules may be required to adequately correct and potentiate the resultant malfunctioning protein.
Investigation of other novel small molecule modulator therapies is underway. For example, the oral potentiator, QBW251, is being evaluated in a phase II trial in adult patients with CF who are heterozygous for mutations resulting in dysfunctional CFTR channels present at the cell surface [ClinicalTrials.gov identifier: NCT02190604]. Additionally, a phase Ib trial has recently been completed using N91115, an oral S-nitrosoglutathione reductase inhibitor shown in vitro to stabilize F508del CFTR [ClinicalTrials.gov identifier: NCT02275936]. Finally, VX-661, an oral CFTR corrector, is being studied in combination with ivacaftor in phase III subjects homozygous [ClinicalTrials.gov identifier: NCT02347657] and heterozygous for the F508del mutation [ClinicalTrials.gov identifier: NCT02392234, NCT02412111].
In addition to evaluation of novel small molecules for CFTR correction, drugs approved for other disease states with potential CFTR activity are being studied. Early phase clinical trials of riociguat (approved for treatment of pulmonary hypertension) and miglustat (approved for type I Gaucher’s and Niemann–Pick disease) are actively recruiting patients with CF homozygous for the F508del CFTR mutation[ClinicalTrials.gov identifier: NCT02170025, NCT02325362].
While the majority of patients may be treated with modulators that effect mutations defective in their folding, gating and conductance, approximately 10% [Peltz et al. 2013] of patients have mutations (‘nonsense mutations’) that result in prematurely truncated CFTR protein. A clinical trial of a therapeutic agent that promotes read-through of premature truncation codons is also enrolling [ClinicalTrials.gov identifier: NCT02139306]. For the remaining 2% of mutations that are unlikely to respond to modulator therapy with small molecules, DNA or RNA editing through oligonucleotide therapy may be an option in the future [McNeer et al. 2015].
Prior to the approval of ivacaftor for patients with the G551D mutation, Mackenzie and colleagues projected that if mortality in CF continued to decrease at the rate observed between 2000 and 2010, predicted median survival for a child born in 2010 would be over 50 years [Mackenzie et al. 2014]. This predicted age is still well below the current survival of the non-CF US population. However, based on the effect of CFTR modulators on FEV1 (our best predictor of mortality in CF) [Kerem et al. 1992], we may rapidly close this survival gap.
Footnotes
Acknowledgements
The first draft of the pharmacokinetics section was written by KK. The remainder of the first draft was written by JTC. JTC revised the final document. Both authors approved the final version of the manuscript. This manuscript has not been submitted or published elsewhere.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: JTC has served as an investigator with grant funding paid to her institution for Vertex Pharmaceuticals Inc., N30 Pharmaceuticals Inc., and Gilead Pharmaceuticals Inc., and has participated in advisory boards for Vertex Pharmaceuticals Inc., Novartis Pharmaceuticals Inc. and Gilead Pharmaceuticals Inc. She has received grant funding paid to her institution from the Cystic Fibrosis Foundation and the National Institutes of Health. KK has no conflicts of interest to declare.