
Abstract
Research over the last decade has determined that the gene rearrangement involving the anaplastic lymphoma kinase (ALK) gene is an oncogenic driver in approximately 5% of patients with non-small cell lung carcinoma (NSCLC). This review describes the discovery of the ALK translocation, development of ALK directed therapy, and acquired resistance to ALK directed therapy with a focus on the clinical data and efficacy of the most recently approved ALK inhibitor, ceritinib.
Keywords
Introduction and background
Non-small cell lung carcinoma (NSCLC) continues to claim more lives than any other cancer each year, while maintaining the status of the second highest incidence of any cancer among both men and women [American Cancer Society, 2014]. Given the challenging nature of this disease and the relatively modest efficacy of current treatment paradigms for the management of advanced stage disease, there has been a great deal of effort put toward the development of novel and targeted treatments. This research strategy has yielded a number of recent advances, including an ever-expanding understanding of the molecular drivers of this disease. The first successfully targeted molecular driver of NSCLC, the activating epidermal growth factor receptor (EGFR) mutations, present in approximately 10% of NSCLC [Marchetti et al. 2005], provided proof of concept that targeted therapy can be of great benefit in appropriately selected patients. This discovery spurred great interest in the EGFR pathway and also generated excitement towards discovering additional molecular drivers of disease that might be similarly targetable with the hope of offering improved disease control and survival. This review examines the discovery of one such genetic event, the activating EML4-ALK (hereafter referred to as ALK) translocation and the development of successful targeted therapy with a focus on the approved second-generation ALK inhibitor, ceritinib.
In 2007, the ALK translocation was described as an oncogenic driver in NSCLC. This gene fusion product between the echinoderm microtubule-associated-protein-like 4 (EML4) and anaplastic lymphoma kinase (ALK) genes, both located on chromosome 2, was demonstrated to give rise to an oncogenic kinase with significantly enhanced activity in vitro and found to be present in approximately 5% of patients with NSCLC [Soda et al. 2007]. In the original description of this ALK translocation, none of the patients were found to be harboring concomitant activating EGFR mutations or KRAS mutations. Soda and colleagues, who identified the ALK translocation in NSCLC, were able to create mouse fibroblast cell lines that expressed the ALK fusion protein in question [Soda et al. 2007]. They then demonstrated that these cell lines injected into mice subcutaneously induced tumor formation, whereas tumors were not formed when wild type fibroblasts alone were injected. Seeking to build on the success of prior TKIs towards oncogene addicted cancers and the evidence of in vitro activity of investigational ALK inhibitors towards inhibiting the growth of ALK+ cultured cell lines [Soda et al. 2007], rapid development of targeted agents was pursued.
Remarkably, only 4 years after the published description of the ALK translocation, crizotinib, an oral small molecule that serves to inhibit the abnormally active ALK fusion protein, received accelerated approval from the US Food and Drug Administration (FDA). Approval was based on phase I data demonstrating a 57% overall response rate in patients who had received prior treatment, a marked improvement over traditional chemotherapeutic second-line therapy [Kwak et al. 2010]. More recently, a phase III trial demonstrated the superiority of crizotinib to second-line chemotherapy (pemetrexed or docetaxel) with an almost 5 month improvement in progression-free survival (PFS) [Shaw et al. 2013]. A subsequent trial established crizotinib as the first-line standard in ALK+ NSCLC with PFS of 10.9 months versus 7.0 months when compared with standard platinum doublet therapy with either cisplatin or carboplatin and pemetrexed [Solomon et al. 2014]. These trials did not demonstrate an overall survival benefit, presumably due to subsequent use of ALK inhibitors among patients who were randomized to the chemotherapy arms. However, they still established the superiority of crizotinib over the previous standard of cytotoxic therapy.
Acquired resistance to crizotinib
Experience to date with approved TKIs for a variety of malignancies has been that cancer cells, over time, develop resistance to therapy by any number of mechanisms including: increasing copy number of the mutated oncogenic driver, mutations that alter drug/target binding and activation of alternative pathways allowing for continued tumor growth. The inevitable emergence of these resistance mechanisms helps explain why the median time to progression on crizotinib is 9 months [Shaw et al. 2013]. Importantly, crizotinib appeared no better at staving off progression than chemotherapy in the central nervous system (CNS) [Solomon et al. 2014]. Owing to this reality, a great deal of research has been undertaken to better understand and catalogue the mechanisms of resistance to first-line ALK inhibition, thereby helping guide the development of second generation inhibitors such as ceritinib.
In hopes of better understanding TKI resistance, molecular analyses of tumor tissues belonging to small cohorts of patients who have progressed on crizotinib have been performed. In one study, 14 patients with ALK+ NSCLC had repeat tissue sampling and molecular analysis [Doebele et al. 2012]. Four of the patients were found to have developed secondary mutations in the tyrosine kinase domain of ALK, thereby interfering with drug binding. Two patients were found to have ALK copy number gain; while three others demonstrated new KRAS mutation or EGFR mutation. The mechanism of resistance for two patients could not be elucidated. A similar study of 18 patients also documented the development of resistance mutations and copy number gain, but two-thirds of the patients did not feature either of these and were thought to be resistant due to over-expression of EGFR or other alternative pathways [Katayama et al. 2012]. These studies and others [Ou et al. 2015] have helped elucidate a number of point mutations that confer resistance including C1156Y, L1196M, L1152R, G1269A, G1202R, S1206Y and an insertion, 1151Tins [Fontana et al. 2015]. The identification of specific mechanisms of resistance raised the tantalizing question as to whether more robust ALK inhibition provided by newer agents could overcome both copy number gain and resistance mutations that disrupt crizotinib’s activity.
Introduction of ceritinib
With the goal of overcoming resistance to first-line ALK inhibition, new ALK inhibitors have been designed in hopes of providing effective salvage therapy. One second generation ALK inhibitor that rapidly generated strong interest was ceritinib (LDK378). This drug was demonstrated to have strong preclinical efficacy in rat xenograft models and notably did not appear to impair glucose homeostasis or confer QT prolongation as crizotinib does in a proportion of patients [Marsilje et al. 2013]. Given encouraging preclinical performance, a phase I escalation/expansion study was undertaken.
The landmark ASCEND-1 study, reported by Shaw and colleagues in March 2014 is the phase I study that first examined ceritinib in cancer patients [Shaw et al. 2014b]. The study enrolled 59 patients in its dose escalation phase and an additional 71 patients in the expansion phase at the time of its initial publication. All patients included in the study had locally advanced or metastatic ALK+ cancer, 94% of which were NSCLC. Prior to enrollment in the study, tumors were required to have 15% of tumor cells ALK+ as evaluated by break apart fluorescence in situ hybridization (FISH). Importantly, a significant majority of patients enrolled, 83 (68%), had already received crizotinib, thus offering the opportunity of studying the drug in both the front line as well as the crizotinib refractory setting.
Toxicity profile of ceritinib
The dose escalation phase of the study determined the mean tolerated dose (MTD) to be 750 mg daily [Shaw et al. 2014b]. Side effects were common with nausea (82% incidence), diarrhea (75% incidence), vomiting (65% incidence), fatigue (47% incidence) and increased alanine aminotransferase levels (35% incidence) of all grades the most common events. The most common grade 3/4 toxicities observed were elevation of the alanine aminotransferase levels (21%), increased aspartate aminotransferase levels (11%), diarrhea (7%) and increased lipase levels (7%). Drug withdrawal led to resolution of abnormalities in all instances. Additional toxicities of note were four possible cases of ceritinib associated interstitial lung disease that all resolved with drug withdrawal and one asymptomatic grade 3 prolongation of the QTc interval. Importantly, at the MTD, 62% of patients required at least one dose reduction. Despite the frequent need for dose reduction, the median duration of treatment interruption was only 1 week and only 6% of patients had study drug discontinued due to adverse event or intolerance. Compared with reported adverse events with crizotinib, visual disturbances, edema, dizziness and constipation were more common with crizotinib and nausea and diarrhea more common in ceritinib. Especially in light of the gastrointestinal (GI) toxicity, ongoing use of ceritinib has required strategies for toxicity management.
Clinical data supporting the efficacy of ceritinib
ASCEND-1 established the efficacy of ceritinib, demonstrating impressive response rates among those studied. The majority of NSCLC patients, 114 in total, received 400 mg doses and above, which all appeared to be similarly efficacious. Patients receiving 400 mg doses and above derived significant benefit with one complete response, 65 partial responses (57%) and 25 (22%) instances of stable disease. Only 12 patients (11%) experienced progressive disease on the initial protocol restaging scan. Impressively, the 83 patients (68%) who had received crizotinib prior to ceritinib had a similar overall response rate (ORR) of 56% to the group as a whole. The ORR among the 34 patients who had never received prior crizotinib was 62%. Among the eight patients who received low doses of ceritinib (300 mg and less), only two partial responses were observed suggesting that these lower doses were insufficient to inhibit ALK strongly [Shaw et al. 2014b].
The findings of the ASCEND-1 study paved the way towards the accelerated approval of ceritinib by the FDA in April 2014 − a rapid introduction and approval of an active second generation drug, only 3 years after the approval of the first line agent, crizotinib.
Updated results after additional accrual to the expansion phase of ASCEND-1, presented at the European Society for Medical Oncology (ESMO) Congress in September 2014 further confirmed ceritinib’s broad activity in ALK+ NSCLC [Felip et al. 2014]. At the time of presentation, data were available for 246 patients with ALK+ NSCLC who received the 750 mg dose during the expansion phase. Median follow up was 7 months. ORR for the cohort as a whole remained similar to initial reports, at 58.5%. Among patients previously treated with crizotinib ORR was 54.6% and in those naïve to ALK directed therapy ORR was 66.3%. Time to response was 6 weeks. Impressively the duration of response (DOR) was 7.4 months for those patients pretreated with a prior ALK inhibitor and 17 months for those ALK directed therapy naïve.
In addition to providing data related to the safety and efficacy of ceritinib, the ASCEND-1 trial featured molecular analysis of ALK mutation among a subset of patients who had progressed on crizotinib prior to enrollment in the study. This subset of 19 patients had tumor biopsy and repeat ALK analysis prior to receipt of ceritinib. Findings were not dissimilar to prior analyses with two patients demonstrating ALK gene amplification and five who had specific resistance mutations. The remaining 12 patients were not identified to have any specific mutations or detectable changes in the ALK domain. Responses were noted in six out of the seven patients with resistance mutation or ALK gene amplification and in seven of 12 patients without identified changes to the ALK domain. These data prospectively demonstrate that ceritinib has efficacy in patients with identifiable mechanisms of crizotinib resistance and in those without such changes identifiable. In the instances where no secondary mutation in ALK is demonstrated by molecular analysis and response to ceritinib is documented, it has been suggested that progression on crizotinib may have been due to crizotinib failing to achieve sufficient drug levels within the tumor tissue, which is then overcome by the more potent drug, ceritinib [Friboulet et al. 2014].
While ALK directed therapy has engendered many dramatic responses systemically, one common site of disease progression has been within the CNS. To date, there has been an increasing understanding that crizotinib does not offer strong CNS penetration and efficacy. There have been a number of reports of patients with excellent responses ongoing to crizotinib who then suffer from progression in the CNS [Chun et al. 2012, Gainor et al. 2013]. Presumably, this is due to poor CNS penetration of crizotinib. This has been physiologically demonstrated in a patient who had cerebrospinal fluid (CSF) and plasma levels measured 5 hours after a dose of crizotinib demonstrating a 237 ng/ml concentration in plasma and only a 0.616 ng/ml concentration in the CSF [Costa et al. 2011]. This had led many to conclude that the CNS is an important sanctuary site for metastatic disease in NSCLC patients who are treated with crizotinib.
Data available within the ASCEND-1 study provide insight into the impact of ceritinib on CNS metastasis. Among the 246 patients in the expansion phase, 124 had brain metastasis and systemic ORR in these patients was 54% with a DOR of 7 months, relatively similar to the group as a whole [Shaw et al. 2014a]. The DOR of patients naïve to ALK directed therapy had not yet been reached at the time of initial presentation and ORR was 69.2%. Among 14 patients (10 ALK pretreated and four ALK naïve), brain metastases were target lesions per Response Evaluation Criteria In Solid Tumors (RECIST) 1.0 criteria. Of these, seven patients (four ALK pretreated and three ALK naïve) had a partial response and three had stable disease, providing evidence that ceritinib has activity and efficacy in the treatment of CNS metastatic disease. Further prospective research is ongoing with a prospective trial for ALK+ patients with CNS progression [ClinicalTrials.gov identifier: NCT02336451].
It is important to highlight that, to date, ceritinib’s indication and approval is restricted to those who have experienced disease progression on or who are intolerant of crizotinib. The optimal sequencing of these two approved ALK directed therapies remains an open question and has yet to have been evaluated prospectively. A trial that is currently underway, the ALEX Study: A Randomized, Phase III Study Comparing Alectinib with Crizotinib in Treatment-Naive Anaplastic Lymphoma Kinase (ALK)-Positive Advanced Non-Small Cell Lung Cancer (NSCLC) Patients is ongoing. While not including ceritinib, it does feature a second generation ALK inhibitor, alectinib, which has been shown in preclinical and clinical studies to also be an active agent [Gadgeel et al. 2014, Kodama et al. 2014]. The primary outcome for this study is PFS with overall survival as a secondary outcome. The results of this study should help begin to offer some guidance as to the optimal timing for initiation of a second generation ALK inhibitor.
Acquired resistance to ceritinib
Resistance mutations to ceritinib and other second generation ALK inhibitors are unfortunately increasingly being described. Mutations including C1156Y, G1202R, 1151T-ins, L1152R and F1174C have been demonstrated in crizotinib-resistant patients and appear to confer varying degrees of resistance to ceritinib as well. Despite this finding, ceritinib remained a more potent inhibitor than crizotinib in both in vitro cell line models as well as murine models with these mutations present [Friboulet et al. 2014]. In a modest cohort of 10 patients with acquired resistance to ceritinib, repeat biopsies were performed with five of the biopsies revealing new mutations at either G1202 or F1174, the two sites that appear to offer the most resistance to ceritinib [Friboulet et al. 2014]. The remaining biopsies, save a single G1269A mutation, demonstrated wild type ALK, suggesting alternative mechanisms of resistance were present in those patients. Other investigation has recently described mutations at V1180L and I1171T that confer resistance to alectinib and crizotinib [Katayama et al. 2014]. These mutations were overcome by ceritinib both in vitro and in vivo, highlighting the fact that second generation ALK inhibitors are not synonymous in their ability to overcome specific point mutations. These findings serve as a reminder that, like crizotinib before them, ceritinib and other second-generation ALK inhibitors will engender the development of resistance necessitating the continued development for additional strategies to overcome these subsequent resistance mutations.
Future directions
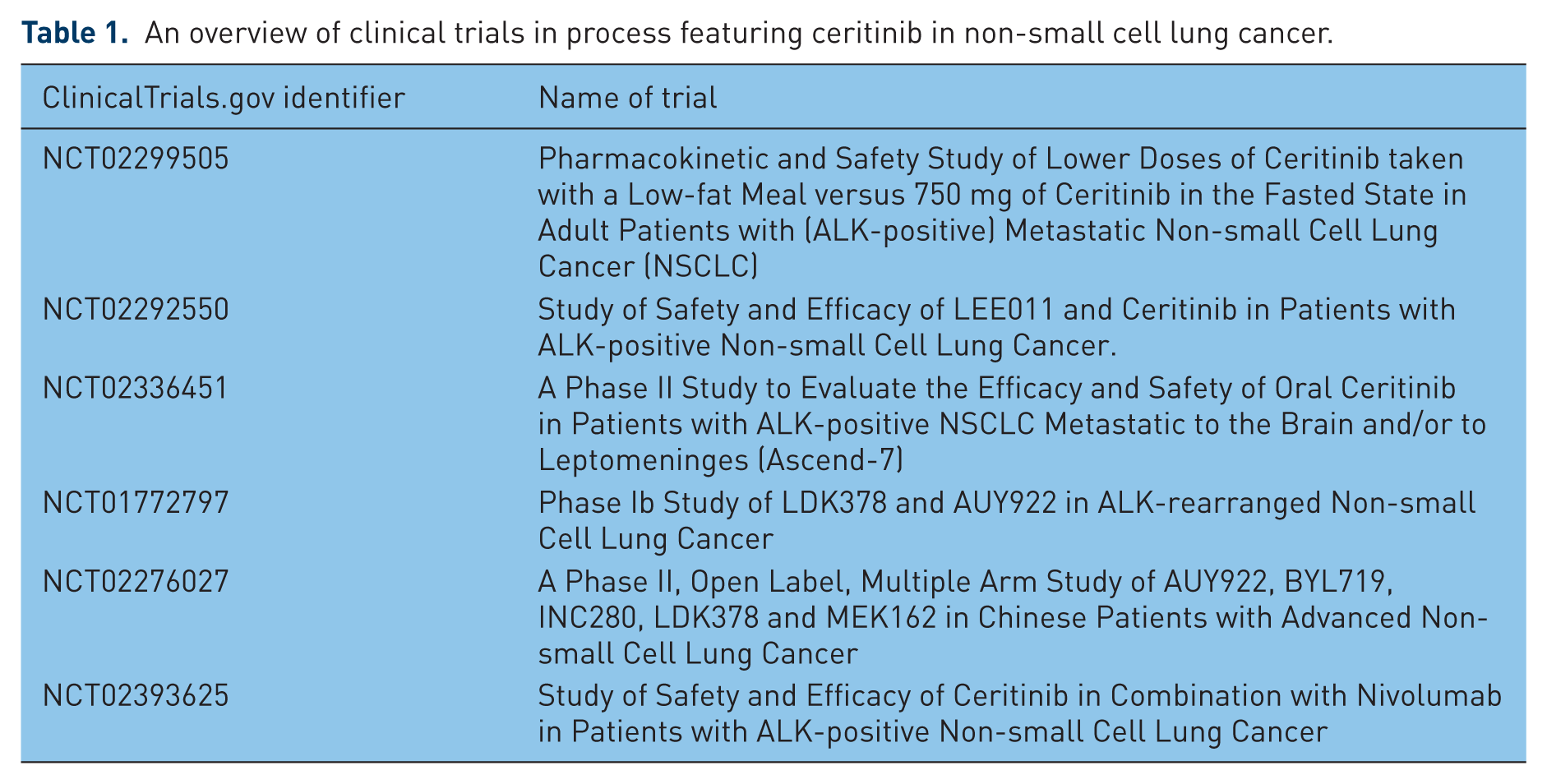
Overall, the treatment landscape for ALK+ NSCLC continues to evolve. The ability of ceritinib to both overcome resistance to crizotinib and control CNS metastasis in a significant proportion of patients offers a valuable addition to the oncologist’s standard armamentarium. With numerous efficacious agents in the pipeline we are seeing the beginning of even more options for ALK+ NSCLC. The contours of this evolution were powerfully demonstrated by a recent report of median overall survival surpassing 4 years in a cohort of 73 patients who received crizotinib followed by ceritinib [Gainor et al. 2015]. Currently, a number of clinical trials are ongoing to explore ceritinib’s performance in combination with other therapies, further characterize its efficacy in CNS disease and explore its dosing and pharmacokinetics in the nonfasting state (Table 1).
An overview of clinical trials in process featuring ceritinib in non-small cell lung cancer.
While undoubtedly resistance to ALK directed therapy will remain a challenge, the increasing hope is that with access to an expanding selection of rationally designed agents, treating oncologists will be able to provide their ALK+ patients with increasingly long periods of disease control and clinical benefit through the use of sequential therapy. The optimal sequencing of such agents and the necessity of repeat biopsy to assess for resistance mutations are two important research questions that will need further clarification. For the time being, two FDA approved agents, crizotinib and ceritinib, provide the ALK+ patient with the hope and the expectation of attaining meaningful periods of disease control and palliation, a truly remarkable achievement in less than 10 years’ time.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.