
Abstract
The use of immunotherapy that harnesses and enhances the innate powers of the immune system to fight cancer cells represents the most promising new cancer treatment approach since the development of the first chemotherapies and, more recently, targeted therapies. Unexpectedly, lung cancer has recently emerged as an exciting new target for immune-based therapies. Several approaches to immunotherapy for lung cancer have shown promise in early clinical trials and in late-phase development. The most advanced strategies can be split into two main categories: therapeutic vaccines and checkpoint inhibitors. At this time of great expectations, this review provides the reader with an update on the immunotherapies used to treat lung cancer with a focus on the rationale of targeting the immune system. It reports the results from recent major clinical trials, describes new toxicity profiles associated with such drugs, and particularly the role of the pulmonologists in their management. This review provides an overview of the main perspectives within this field.
Keywords
Introduction
Activation of the immune system to treat cancer has long been investigated. After decades of disappointment, the tide has now finally changed due to the success of recent phase I clinical trials, especially for diseases with limited therapeutic options, such as melanoma and lung cancer. Durable responses have been recently reported for a broad range of human cancers using different agents that target the immune checkpoints [Gettinger and Herbst, 2014; Hamid and Carvajal, 2013; Hodi et al. 2010; Topalian et al. 2012]. In addition to their promising activities, these treatments are usually well tolerated.
We are currently in a transition period in treating lung cancer with immunotherapy: there are encouraging phase I–II trials, and numerous ongoing phase III clinical trials, but only one drug gets a recent approval in the USA. In this time of great expectations, we have aimed for this review to provide the reader with an update on the use of immunotherapy to treat lung cancer, with a focus on the rationale of targeting the immune system. We describe results obtained from major clinical trials, the new toxicity profiles associated with such drugs, and particularly the role of pulmonologists in their management. We also provide a comprehensive overview of the main perspectives within this field.
Rational for targeting the immune system in lung cancer
It is somehow unexpected that, besides melanoma, the most promising results with immunotherapy to treat cancer have been observed in lung cancer. We argue that many features of lung cancer support these findings (Figure 1).

Rationale for targeting the immune system in lung cancer. (a) Example of spontaneous regression of lung cancer [Gladwish et al. 2010]. (b) Example of usual interstitial pneumonia showing the frequency of interstitial immune-related disease of the lung. (c) The thin interface between the bronchoalveolar compartment and intrinsic immune regulators. (d) PDL1 expression in lung cancer. PDL1, programmed cell death ligand 1.
Legend of table 2.
First, the lungs are the sanctuary of many immune disorders, along with the skin and kidneys (interestingly, all three organs being the best candidates for anticancer immunotherapy). Most autoimmune pathologies, such as disseminated lupus, Wegener’s disease, and other vascularitis are associated with lung complications. Pulmonary fibrosis is also a frequent disease linked to immune escape. Even more frequent diseases, such as asthma or chronic obstructive pulmonary disease, are associated, at least in part, with immune disorders, such as an imbalance in the T helper type 1 (Th1)/Th2 response.
Second, the lungs are rich in various types of immunity cells, probably due to their chronic exposure to numerous external agents and the subsequent inflammation caused [Dasanu et al. 2012]. Among these cells, regulatory T cells have been shown to play a major role in immune surveillance and immune adoptive responses.
Third, the unique interface between the bronchoalveolar compartment and intrinsic immune regulators may play an important role in facilitating inflammatory alterations that promote lung cancer. Lung oncogenesis is marked by a complex array of immune defects, such as important T-cell alterations, natural killer cell dysfunction, B-lymphocyte defects, and defective dendritic cell, neutrophil, and monocyte functions [Sterlacci et al. 2012; Wesselius et al. 1987; Zikos et al. 2011].
Fourth, antigens are frequently expressed in lung tumor cells, some of which are shared with normal tissues, whereas others are unique to tumors. Tumor-specific antigens, such as angiotensin-converting enzyme, KRas, p53, and human telomerase reverse transcriptase, are frequently overexpressed or mutated in lung cancer [Hilbe et al. 2004] and are known to induce an immune response.
Lastly, some clinical reports of spontaneous resolution of lung cancer (as observed in patients with melanoma) suggest the importance of the immune response in tumor control [Gladwish et al. 2010].
Main mechanisms
The main research findings supporting the use of immunotherapy in cancer are extensively reported in very elegant reviews [Chen and Mellman, 2013; Mellman et al. 2011]. We focus here on the steps of the cancer immunity cycle that can be targeted in lung cancer and the main mechanisms set up by the tumor to escape the immune response. Immunotherapy is considered active when it acts directly on the immune system and passive when its antitumor activity is based on an immunological mechanism.
Active immunotherapy
Active immunotherapy in oncology can be split into two main categories: those whose antitumor activity is antigen dependent and those that target immunological checkpoints. Treatments based on cytokines to enhance antitumor immune response are just mentioned here.
Antigen-dependent immunotherapy
The purpose of the tumor-associated antigen (TAA) vaccines is to promote an antitumor immune response by enhancing the presentation of TAAs by dendritic cells to naïve T cells Major histocompatibility complex/T cell receptor (MHC–TCR interaction). This is the most widely studied immunotherapy. Yet the results are still disappointing (see below for details of the clinical studies). This is due to the different barriers related to the complexity of the immune response. The absence of dendritic cells that activate an adjuvant is one of the main reasons: the best adjuvant has still not been determined [granulocyte macrophage colony-stimulating factor (GM-CSF) and interleukin 2 (IL2) being the most widely used]. Determination of the best TAA candidate appears to be very tricky.
The ideal antigen should be uniformly expressed on tumor cells but be missing from normal cells. It is not yet established whether the vaccine should be monovalent (with the risk of secondary resistance antigenic escape) or plurivalent. Another limiting factor is that the antigen may not be recognized or be recognized as a self antigen and thus induce a tolerogenic response [Motz and Coukos, 2013]. The tumor cell may also evade the immune system through the loss of expression of tumor antigens or MHC [Marincola et al. 2000; Whiteside, 2010]. Lastly, immune surveillance or artificial antigen-based immunotherapy can lead to selective survival of tumor cells that lack immunogenic epitopes, a process known as immunoediting [Schreiber et al. 2011].
Antigen-independent active immunotherapy: immune checkpoint blockade
Immune checkpoint mechanisms normally prevent excessive and uncontrolled immune responses [Nirschl and Drake, 2013]. Briefly, TAAs must be directly presented by tumor cells or captured, processed, and presented by dendritic cells. These latter can, in turn, differentiate, migrate to the lymph nodes, and present TAA to naive T cells. The next step involves expansion of T cells in sufficient numbers to recognize and eliminate tumor cells. Antigen-educated T cells then leave the lymph node, travel to infiltrate the tumor, and eventually persist for enough time to kill the malignant cells. Immune checkpoints are crucial in this tumor immunity cycle and thus constitute potential targets for anticancer drugs. The priming of T-cell activation can be targeted by anticytotoxic T lymphocyte associated protein 4 (CTLA4) antibodies, such as ipilimumab or tremelimumab, which block the interaction of the major negative regulator of T cells (CTLA4) with its ligands B7.1 and B7.2 (CD80 and CD86) [Qureshi et al. 2011]. CTLA4/CD80-86 interaction leads to anergy or redirects the T cells to a tolerogenic phenotype CD25-Foxp3 (T-regulatory cells), representing an ideal way for the tumor to escape the immune system [Darrasse-Jèze et al. 2009; Steinman et al. 2000, 2003]. The identification of programmed cell death ligand 1 (PDL1) as a distal immune modulator, expressed in 20–50% of human cancers [Gettinger and Herbst, 2014], has also led to the development of a number of cancer immunotherapies that target the interactions between PD1 (programmed cell death 1) and PDL1, PDL1/B7.1, and PDL2/PD1 [Chen et al. 2012; Topalian et al. 2012].
Anti-CTLA4 monoclonal antibodies thus enable activation and expansion of tumor-infiltrating lymphocytes with a cytotoxic antitumor phenotype. This strategy’s main disadvantage is its obvious lack of selectivity and, therefore, the potential expansion of autoreactive lymphocytes that can induce autoimmune toxicity [Fong and Small, 2008].
The PDL1/2/PD1 pathway is a more distal secondary inhibitory immune checkpoint [Chen, 2004]. The transmembrane molecule PD1 is found on the surface of T lymphocytes, B lymphocytes, and monocytes [Keir et al. 2008]. PDL1 is expressed either on tumor cells or on tumor-infiltrating immune cells. Their interaction causes the recruitment of the phosphatase Src-Homology Protein 2 (SHP2), subsequent inactivation of the PI3K/AKT signaling cascade, a change in the T-cell transcriptional profile, including blocking the secretion of cytotoxic mediators [Parry et al. 2005]. This pathway represents another immune escape mechanism for the tumor [Taube et al. 2012].
Passive immunotherapy: dendritic-cell-based immunotherapy
Dendritic cells may acquire different phenotypes: proinflammatory when they secrete IL15, inducing a cytotoxic CTL response, and conversely more tolerogenic when they secrete interferon α (INFα) to induce a tolerogenic protumoral response mediated by INFγ and IL10 secretion [Banchereau and Palucka, 2005].
Proinflammatory dendritic cells can be generated ex vivo from patients’ monocytes, loaded with TAAs, and then activated and injected into patients [Palucka and Banchereau, 2013]. Dendritic-cell-based vaccines induce expansion of circulating CD4+ and CD8+ T cells, which are specific for tumor antigens.
Passive immunotherapy: adoptive transfer of ex-vivo-activated T cells
Chimeric antigen receptors are composed of a synthetic antibody against a tumor-surface antigen (with CD19 for B-cell malignancies being the most widely studied) fused to T-cell signaling domains [Kalos and June, 2013]. Genetically modified autologous transfected T cells are then reinfused into patients [Kochenderfer and Rosenberg, 2013]. This strategy still needs to be extended to cancers beyond hematologic malignancies.
Vaccines: clinical results
Antigen-specific active immunotherapies have been tested in several clinical trials with, up to now, disappointing results. Most antigen-specific immunotherapies have incorporated a single antigen and have narrow epitope specificities, which may contribute to their lack of efficacy in some trials [Slingluff, 2011]. We briefly report on the main trials below.
Targeting MUC1 (Mucin 1)
The phase III START trial [Butts et al. 2014] randomized more than 1500 patients with unresectable stage III non-small cell lung cancer (NSCLC), previously treated with chemoradiotherapy, between treatment with tecemotide (a therapeutic vaccine targeting cancer cells expressing MUC1) or a placebo. Tecemotide failed to significantly prolong survival in the overall population. However, in a preplanned subgroup analysis, tecemotide improved survival in patients who had received concurrent chemoradiotherapy [30.8 months versus 20.6 months; hazard ratio (HR) 0.78, p = 0.016]. A further phase III trial is being initiated in this population (START2). The TG4010 vaccine also targets MUC1, but consists of a recombinant vaccinia virus encoding MUC1 and IL2. In a phase II study on advanced stage NSCLC, a slight albeit nonsignificant difference in progression-free survival (PFS) at 6 months was observed (43% with TG4010 combined with chemotherapy versus 35% with chemotherapy alone, p = 0.3) and no difference in terms of overall survival (OS) (10.7 months versus 10.3 months; p = 0.59). A phase IIB–III study of TG4010 added chemotherapy to stage IV NSCLC (TIME trial): it has recently shown that TG4010 was efficacious and had an acceptable safety profile in patients with stage IV NSCLC, particularly in the nonsquamous population [Vansteenkiste et al. 2013; Quoix et al. 2014]. Moreover, exploratory analysis of lymphocyte phenotypes at baseline performed in patients with evaluable samples suggested that the percentage of CD16+CD56+CD69+ cells, a phenotype of activated natural killer cells, was a potential predictor of outcome in patients who received TG4010 [Quoix et al. 2011].
Targeting melanoma-associated antigen 3
GSK1572932A is a vaccine that combines a melanoma-associated antigen 3 (MAGE-A3) peptide with the immune adjuvant AS15. A phase II study in patients with resected MAGE-A3+ NSCLC initially suggested a trend towards improved outcomes [Vansteenkiste et al. 2013; Quoix et al. 2014]. Unfortunately, these results were not confirmed in a recent large phase III study (MAGRIT trial), which showed no difference in terms of disease-free survival and no clear impact of the biomarkers [Vansteenkiste et al. 2014].
Targeting human telomerase reverse transcriptase
GV1001, a peptide vaccine that corresponds to the active site of human telomerase reverse transcriptase and GM-CSF, induced specific immune responses in 80% of patients with unresectable stage III NSCLC [Brunsvig et al. 2011]. A gain in median OS was reported among immune responders (19.0 months versus 3.5 months for immune nonresponders, p < 0.001) in a phase I/II study in which GV1001 was combined with a second telomerase peptide (I540). A phase III study is ongoing.
Other strategies
The CIMAvax epidermal growth factor vaccine, developed in Cuba, is designed to induce an antibody-mediated immune response [Vinageras et al. 2008]. However, a phase II study failed to demonstrate a gain in median OS, but a subgroup of patients with a good antibody response appeared to derive some benefit from the drug (11.7 months versus 3.6 months, p = 0.002).
The belagenpumatucel-L vaccine (LucanixTM, NovaTx Corporation, San Diego, California, USA) consists of NSCLC cell lines transfected with a transforming growth factor β2 antisense gene. Despite a promising phase II study [Nemunaitis et al. 2006], a recent phase III trial on advanced-stage NSCLC reported no significant difference in OS compared with a placebo in the overall study population, but with a potential benefit in subgroups with adenocarcinoma and who had been previously treated with radiotherapy [Giacone, 2013].
Tergenpumatucel-L (lung-cancer cell lines transfected with a murine galactosyl-transferase gene) was associated with median OS of 11.3 months in a phase II study that included 28 patients with metastatic or recurrent NSCLC. Induction of IFNγ secretion by antigen-specific T lymphocytes was associated with improved OS (21.9 months versus 7.2 months, p = 0.044) [Morris et al. 2013].
Altogether, the results are disappointing to date but subgroups appeared to derive more benefit than other groups and new trials are currently being conducting in selected patients.
Targeting CTLA4
CTLA4 expression is not only confined to T lymphocytes but is also expressed in NSCLC tumors in 51–87% of cases. In one study, CTLA4 was associated with nonsquamous histology but was not prognostic for OS [Salvi et al. 2012]. In another study, CTLA4 tumor expression was associated with older age and poor tumor differentiation [Sundar et al. 2014; Zheng et al. 2010].
Ipilimumab
Ipilimumab is an anticytotoxic T-cell lymphocyte 4 monoclonal antibody that was approved, in 2011 by the US Food and Drug Administration (FDA) to treat all patients with unresectable or metastatic melanoma. It was also approved, in 2013, by the European Medicines Agency for adult patients with previously treated advanced melanoma. Ipilimumab has been evaluated in a stage IIIb/IV NSCLC phase II study in combination with paclitaxel and carboplatin. Patients were randomized to receive either paclitaxel with carboplatin and a placebo, or concurrent ipilimumab or phased ipilimumab (frontline chemotherapy alone and subsequent combination of both regimens). Phased ipilimumab improved the immune-related PFS (5.7 months versus 4.6 months with a placebo) (HR 0.72, p = 0.05) and median OS (12.2 months versus 8.3 months with a placebo), but the difference in OS was not significant (p = 0.23). The PFS advantage was not observed in the concurrent ipilimumab arm [Lynch et al. 2012].
Two phase III studies [ClinicalTrials.gov identifier: NCT01285609 and NCT01450761] have recently evaluated ipilimumab in combination with paclitaxel–carboplatin or platinum–etoposide in patients with squamous NSCLC and small cell lung cancer. Results are awaited. Other trials are currently ongoing, such as a two phase I trials on ipilimumab plus targeted therapies for patients with stage IV NSCLC with epidermal growth factor receptor (EGFR) or ALK mutations [ClinicalTrials.gov identifier: NCT01998126]. Another trial is assessing ipilimumab plus imatinib [ClinicalTrials.gov identifier: NCT01738139], and a phase II neoadjuvant trial is assessing ipilimumab plus chemotherapy [ClinicalTrials.gov identifier: NCT01820754].
A second antibody against CTLA4, tremelimumab, is a human immunoglobulin G2 (IgG2) monoclonal antibody. This antibody has been studied in a phase II open-label trial on advanced malignant mesothelioma in which the patients were chemotherapy resistant. In the tremelimumab arm, the median PFS was 6·2 months [95% confidence interval (CI) 1.3–11.1] and the median OS was 10.7 months (95% CI 0.0–21.9). A phase IIb, randomized, double-blind study that is comparing tremelimumab with a placebo for the second- or third-line treatment of subjects with unresectable pleural or peritoneal malignant mesothelioma is ongoing.
Targeting the PD1/PDL1 pathway
PDL1 is a distal modulator of the immune response whose expression occurs in 40–50% of NSCLC cases [McLaughlin et al. 2014; Yang et al. 2014]. Its prevalence is higher in men with squamous cell carcinoma at an advanced stage [McLaughlin et al. 2014]. Although no association has been established with the main molecular biomarkers in stage I resected lung adenocarcinoma [Yang et al. 2014], recent data suggest a correlation between tobacco and the KRAS mutation (OR 2.5) among all stages of combined populations [Ansen et al. 2014; Calles et al. 2014]. The main molecules currently under investigation are summarized in Table 1. The outcomes and toxicities observed with different molecules targeting PDL1/PD1 pathways are summarized in Table 2. Figure 3 shows the analysis of PDL1 expression according to companies developing checkpoint inhibitors. Figure 4 shows two individual cases of tumor responses and toxicities after treatment with anti-PDL1 inhibitors.
Overview of the main checkpoint inhibitors developed in lung cancer

CTLA4, cytotoxic T lymphocyte associated protein 4.
Efficacy and toxicity observed with the main checkpoint inhibitors in lung cancer.

AE, adverse event; NR, not reported; OS, overall survival; PD1, programmed cell death 1; PDL1, programmed cell death ligand 1; PFS, progression-free survival; RR, relative risk.

Algorithm of management pulmonary symptoms in patient treated with immunotherapy.

Example of tumor response after treatment with programmed cell death ligand 1 inhibitor (a, b) and example of programmed cell death 1 inhibitor induced interstitial pneumonia (c) (all personal cases).
Anti-PD1
Nivolumab, an anti-PD1 IgG4 monoclonal antibody, is the first and most extensively evaluated anti-PD1 inhibitor of NSCLC. Results from a phase Ib study on nivolumab have reported objective response rates for pretreated NSCLC of 17% [Topalian et al. 2012]. Median OS was 9.9 months, and was uninfluenced by histology (9.2 for squamous cell histology and 10.1 for a nonsquamous histology). It is important to note that efficacy appears sustainable (74 weeks median response duration, 1-year OS 42%, 2-year OS 24%) [Brahmer et al. 2014]. The best results were obtained at the dose of 3 mg/kg (1-year OS 50%, 2-year OS 45%) [Brahmer et al. 2014].
A recent update shows a 3-year survival rate of 17% for all patients and 27% for patients receiving the 3 mg/kg dose [Gettinger and Herbst, 2014]. Given frontline as a monotherapy, at a dose of 3 mg/kg every 2 weeks, a response rate of 21% was observed, and this was higher in smokers and in patients harboring expression of PDL1 [Rizvi et al. 2014]. Median PFS was 15.6 weeks. As a first-line therapy, at a dose of 5 mg/kg, disease control rate was 51%, 71%, and 38%, when combined with gemcitabine–cisplatin, pemetrexed–cisplatin, and paclitaxel–carboplatin, respectively [Scott et al. 2014].
A recent phase II study (CA209-063) was conducted in pretreated patients with squamous-cell lung carcinoma [Rizvi et al. Lancet Oncol 2015]. Nivolumab, as a monotherapy, showed clinically meaningful efficacy in this population with an overall response rate (ORR) of 15%, durable responses (59% ongoing), and a 1-year OS of 41%. Clinical activity was observed in both PDL1– and PDL1+ patients with a higher although not significant benefit for the PDL1+ patients (ORR 24% versus 14%). Nivolumab was well tolerated with a manageable safety profile, but 12% of patients discontinued treatment due to drug toxicity (4% pneumonitis), and two treatment-related deaths (one hypoxic pneumonia and one ischemic stroke) occurred in patients with multiple comorbidities and concurrent progressive disease. Two phase III trials that compared OS with nivolumab versus docetaxel in advanced, previously treated nonsquamous (CA209-057) [ClinicalTrials.gov identifier: NCT01673867] and squamous NSCLC have been recently completed (CA209-017) [ClinicalTrials.gov identifier: NCT01642004].
Pembrolizumab (MK-3475) is another IgG4 anti-PD1 antibody. Pembrolizumab was approved by the FDA in September 2014 for the treatment of advanced melanoma and was granted a ‘breakthrough-therapy’ designation in October 2014 for the treatment of patients with pretreated NSCLC who were negative for EGFR and ALK. This study follows the encouraging intermediary results of the KEYNOTE-001 study (n = 307), which showed a response rate of 21% (RECIST) and 23% (immune-related response criteria) [Wolchok et al. 2009]. A median PFS of 27 months among treatment-naïve patients and 10 months among pretreated patients was observed. Again, the responses observed were usually durable [Garon et al. 2014]. Using a stringent test, with a positivity cutoff at 50% of cells expressing PDL1, a clinically relevant difference was observed in terms of response between the strong positive group (37%) and the negative group (10%).
First-line and combination trials are ongoing, as follows. There is a phase III trial on pembrolizumab given to patients with PDL1+ NSCLC [ClinicalTrials.gov identifier: NCT01905657], a phase II study on pembrolizumab given to patients with NSCLC and brain metastases [ClinicalTrials.gov identifier: NCT02085070], a phase I/II trial on pembrolizumab in combination with chemotherapy or a targeted therapy for patients with NSCLC [ClinicalTrials.gov identifier: NCT02039674], a phase I/II trial on pembrolizumab in combination with an IDO (indoleamine 2,3-dioxygenase) inhibitor given to patients with advanced NSCLC [ClinicalTrials.gov identifier: NCT02178722], and a phase I trial on pembrolizumab given to patients with biomarker-positive solid tumors, including lung cancer [ClinicalTrials.gov identifier: NCT02054806].
Anti-PDL1
MPDL3280A, a human anti-PDL1 monoclonal antibody, led to antitumor responses, with an ORR of 23% (12/53) in patients with previously treated NSCLC in a phase I study, with a rapid and durable response [Figure 4(a, b)] [Brahmer 2014]. Moreover, the treatment was well tolerated. Patients with PDL1+ tumors showed an ORR of 39%, and a progressive disease rate of 12%. Patients with PDL1– tumors had an ORR of 13% with a progressive disease rate of 59%. These results suggested a correlation between a response to the agent and PDL1 status [Herbst et al. 2013; Spiegel et al. 2013].
Other one-arm or randomized phase II and III studies are ongoing. Following a phase II randomized trial (POPLAR) [ClinicalTrials.gov identifier: NCT01903993], a phase III trial on MPDL3280A (OAK) [ClinicalTrials.gov identifier: NCT02008227] has just been completed. This open-label, multicenter study is designed to evaluate the efficacy and safety of MPDL3280A versus docetaxel in patients with locally advanced or metastatic NSCLC who have failed a platinum-based chemotherapy. Two phase II trial are dedicated to patients with PDL1+, locally advanced or metastatic NSCLC (FIR and BIRCH) [ClinicalTrials.gov identifier: NCT01846416, NCT02031458]. A phase I trial is evaluating the combination of MPDL3280A combined with bevacizumab or bevacizumab plus chemotherapy for patients with advanced NSCLC [ClinicalTrials.gov identifier: NCT01633970].
MEDI4736 is another anti-PDL1 monoclonal antibody that achieved a 13% objective response rate and a 30% disease control rate in NSCLC, in a phase I study [Segal et al. 2014]. Currently within recruitment, the ATLANTIC [ClinicalTrials.gov identifier: NCT02087423] is a single-arm, phase II global study on the efficacy and safety of MEDI4736 in pretreated patients with locally advanced or metastatic NSCLC. The PACIFIC trial [ClinicalTrials.gov identifier: NCT02125461] is a phase III study on MEDI4736 versus a placebo given to patients with locally advanced, unresectable NSCLC, with no evidence of progression after completion of treatment of chemoradiotherapy. A phase II/III trial on MEDI4736, as a second-line therapy, is also ongoing for patients with recurrent stage IIIB/IV NSCLC [ClinicalTrials.gov identifier: NCT02154490]. In addition, there are several early phase I studies on NSCLC using MEDI4736 in combination with gefitinib [ClinicalTrials.gov identifier: NCT02088112], tremelimumab [ClinicalTrials.gov identifier: NCT02000947], and MEDI0680 [ClinicalTrials.gov identifier: NCT02118337], an anti-PD1 monoclonal antibody (in advanced malignancies).
The different drugs targeting the CTLA4 and PD1/PDL1 pathways and the ongoing phase II/III clinical trials are summarized in Table 1.
Toxicity
Targeting immune checkpoints induced the emergence of a new form of toxicity termed immune-related adverse effects (irAEs). Due to its more distal localization in the cancer immunity cycle, inhibition of the PD1 pathway enhances the effector function of previously activated T cells. Conversely, CTLA4 blockade both promotes the activation of preexisting T cells and redirects naïve T cells into the cytotoxic phenotype. This difference theoretically suggests a lower risk of autoimmune disorders by targeting the PD1/PDL1 pathway. Anti-PDL1 antibodies may also have better selectivity for tumor cells than anti-PD1, with fewer irAEs [Zou and Chen, 2008].
The most commonly observed irAEs with all checkpoint inhibitors are the following:
Lung: pneumonitis is defined as a disorder characterized by focal or diffuse inflammation that affects the lung parenchyma.
Skin: rash or pruritus (grade 1) is frequent. The appearance of a rash may be explained by an ipilimumab-stimulated immune response to melanocytes [Weber et al. 2009]. This is supported by the appearance of vitiligo in 11% of patients in a phase II study on ipilimumab [Hodi et al. 2003]. Toxic epidermal necrolysis, as well as Stevens–Johnson syndrome (grade 3–4), are rarely reported.
Gastrointestinal: diarrhea and colitis are frequent, whereas hepatotoxicity and pancreatitis are uncommon.
Endocrine glands: hypophysitis and hypothyroidism have been reported.
Anti-CTLA4
Blockade of CTLA4 signaling with ipilimumab or tremelimumab increases T-cell activation and restores T-cell proliferation. Part of the toxicity (irAEs) is probably induced by cytokines released by activated T cells. In a pooled analysis of 325 patients treated with 10 mg/kg ipilimumab, once every 3 weeks for four times: irAEs of any grade were observed in 72.3%. Grade 3 or 4 irAEs were observed in 25.2% of patients, mainly in the gastrointestinal tract (12%), liver (7%), skin (3%), and endocrine system (3%) [Weber et al. 2012].
Anti-PDL1
IrAEs, including mild fatigue, rash, diarrhea, and colitis, have been described with anti-PD1 agents, although adverse events seem to be less common than those seen with anti-CTLA4 antibodies [Gangadhar and Vonderheide, 2014]. Nivolumab was generally well tolerated with skin toxicities (20%), gastrointestinal (15%), and pulmonary (9%) adverse events being most commonly observed. A lower frequency of gastrointestinal toxicities was seen: 2% (grade 3/4) compared with 20% with ipilimumab. Pneumonitis was reported in 6% (8/129) [Brahmer et al. 2014]. A rare but potentially fatal inflammatory pneumonitis was observed in a few cases [Figure 4(c)], which led to three treatment-related deaths in the original phase I study on nivolumab [Topalian et al. 2012] and one related death in the CHECKMATE 063 trial [Rizvi et al. Lancet Oncol 2015]. The other drugs, pembrolizumab, MEDI-4736, and MPDL3280A, have very similar safety profiles with mild pulmonary toxicities. The main toxicities observed with checkpoint inhibitors are summarized in Table 2.
Management of pulmonary toxicity
Pulmonary toxicity needs to be recognized and treated early as it can be life threatening. Pneumonitis management has been described in clinical experience and observational reports but without consensus, standardization, or validation in prospective clinical trials [Inoue et al. 2001; Lynch et al. 2012]. It is unknown whether immune-related pneumonitis relates to other forms of described pneumonitis or how it should best be managed [Chow, 2013]. Other forms of severe or even fatal drug-related pneumonitis have been described in patients with NSCLC treated with erlotinib (1.6–4.5%) [Liu et al. 2007], gefıtinib (3.5%) [Konishi et al. 2005], docetaxel (4.6%) [Grande et al. 2007], and gemcitabine (1–2%) [Roychowdhury et al. 2002], with two case reports of pemetrexed-related pneumonitis [Hochstrasser et al. 2012].
Patients with signs and symptoms of pneumonitis, such as dyspnea, cough, hypoxia, and interstitial syndrome (as observed on a chest X-ray) should undergo a computed tomography scan. This exam can confirm pneumonitis (extensive, patchy, bilateral, peribronchial consolidation) or orient towards a differential diagnosis (infection, heart failure, tumor progression, pulmonary embolism). If there is any doubt, a bronchoscopy should be considered with an oriented bronchoalveolar lavage that includes a cell count, analysis of T-cell subsets, microbial analysis (including viruses, fungi, bacteria and BK) and, according to the patient’s status, a transbronchial biopsy, which can help narrow down the diagnosis [Barjaktarevic et al. 2013; Rizvi et al. Lancet Oncol 2015]. Some recommendations have been proposed [Ramaligan et al. 2014] based on the following clinical signs.
For grade 1 (asymptomatic, clinical, or diagnostic observations only, intervention not indicated): it is possible to discuss utilization of steroids and empiric antibiotics (if there is a suspicion of concurrent infection) and to repeat thoracic imaging 3 weeks later.
For grade 2 (symptomatic and medical intervention is indicated, limited ability for daily living): delay immunotherapy dosing and start moderate doses (1–2 mg/kg) of prednisone, which is then slowly tapered over 4 weeks or more; a bron-choalveolar lavage with a bronchoscopy is also recommended.
Grade 3 (severe symptoms, limited self care or activities of daily living, or oxygen) and above, immunotherapy treatment should be discontinued permanently and high doses of intravenous steroids (for example: methylprednisolone 1 g/day) administered in conjunction with oxygen and ventilatory support, if needed. Bronchoscopy or lung biopsies are indicated if clinically feasible. In cases of failure after 48 h or serious evolution, it should be considered to use broad-spectrum prophylactic antibiotics and additional immunosuppressive medication. In the protocol of some clinical trials addition of infliximab, mycophenolate mofetil, or cyclophosphamide is proposed even if data are lacking to validate the use of such drugs. Herein, we propose an algorithm to help manage the onset of pulmonary symptoms in patients treated with immunotherapy (Figure 3).
Perspectives
Combination of drugs
Even though some immunotherapies may have activity as single agents, more impressive activity can be seen when they are combined with other agents. Combinations of immune therapies and chemotherapy agents are currently being studied in the setting of advanced disease. Accumulating evidence suggests the rational for different drug associations:
Chemotherapies and immunotherapies
Several conventional chemotherapies that induce immunogenic tumor-cell death can enhance a strong adaptive immune response. Indeed, tumor cells are converted into an anticancer vaccine (release of endogenous tumor antigens), and their effects may be enhanced by immune checkpoint inhibitors [Vacchelli et al. 2014]. Numerous clinical trials are ongoing, and are testing such combinations in small cell lung cancer and NSCLC.
Anti-PD1/PDL1, anti-CTLA4 antibodies, and antitumor vaccines
Prolonged antigenic exposure leads to expression of PD1/PDL1 and T-cell anergy [Blackburn et al. 2009]. This constitutes a robust rational for a synergic combination of antigen-specific active immunotherapies with immune checkpoint inhibitors because CTLA4 and PD1 inhibit T-cell activation and clonal T expansion after vaccination.
Anti-PD1/PDL1 and anti-CTLA4 antibodies
Because of two distinct levels of intervention, that is, T-cell activation and expansion in the periphery of CTLA4 and CTL effector functions in the tumor microenvironment of PD1, it seems logical to combine these two drugs to enhance antitumor immune activity [Wolchock et al. 2013]. The combination of ipilimumab plus nivolumab has shown response rates of 11–33% depending on histology, but induces frequent and sometimes severe irAEs (85% toxicity, 48% grade 3–4, three deaths among 46 patients) [Antonia et al. 2014].
Immunotherapy and targeted therapy
Activating a patient’s immune system during a time of tumor reduction and remission may be the best way to ensure that responses are converted to a long-term and durable benefit. Unlike conventional chemotherapies, targeted therapies for EGFR-mutated or ALK-rearranged adenocarcinoma may achieve rapid and significant tumor shrinkage without the need for immunosuppression induced by chemotherapies. Moreover, oncogenic EGFR signaling remodels the tumor microenvironment to trigger an immune escape and mechanistically links the treatment response to PD1 inhibition [Akbay et al. 2013]. A first-line therapy that combines nivolumab and erlotinib shows excellent response rates, even in EGFR–tyrosine kinase inhibitor (TKI) pretreated patients, but with a grade 3–4 adverse event incidence of 24% [Naiyer et al. 2014]. Other trials combining, for example, gefitinib and tremelimumab in patients with EGFR mutation who progress under EGFR–TKI are ongoing.
Biomarkers
As with other targeted therapies, only a minority of patients currently benefit from active immunotherapy even though, for some patients, the benefits can be substantial. Patient selection is therefore a crucial issue in the development of such drugs. Initial assessments of active immunotherapies have generally been in patients with advanced-stage disease. However, rapid tumor growth and extensive tumor-related immunosuppression suggest that these patients may be least likely to benefit from this treatment.
Ipilimumab
Among patients treated with ipilimumab, an early increase in lymphocyte and eosinophil counts is associated with improved survival but no robust pretreatment biomarker has yet been identified [Delyon et al. 2013].
Anti-PD1 and anti-PDL1 antibodies
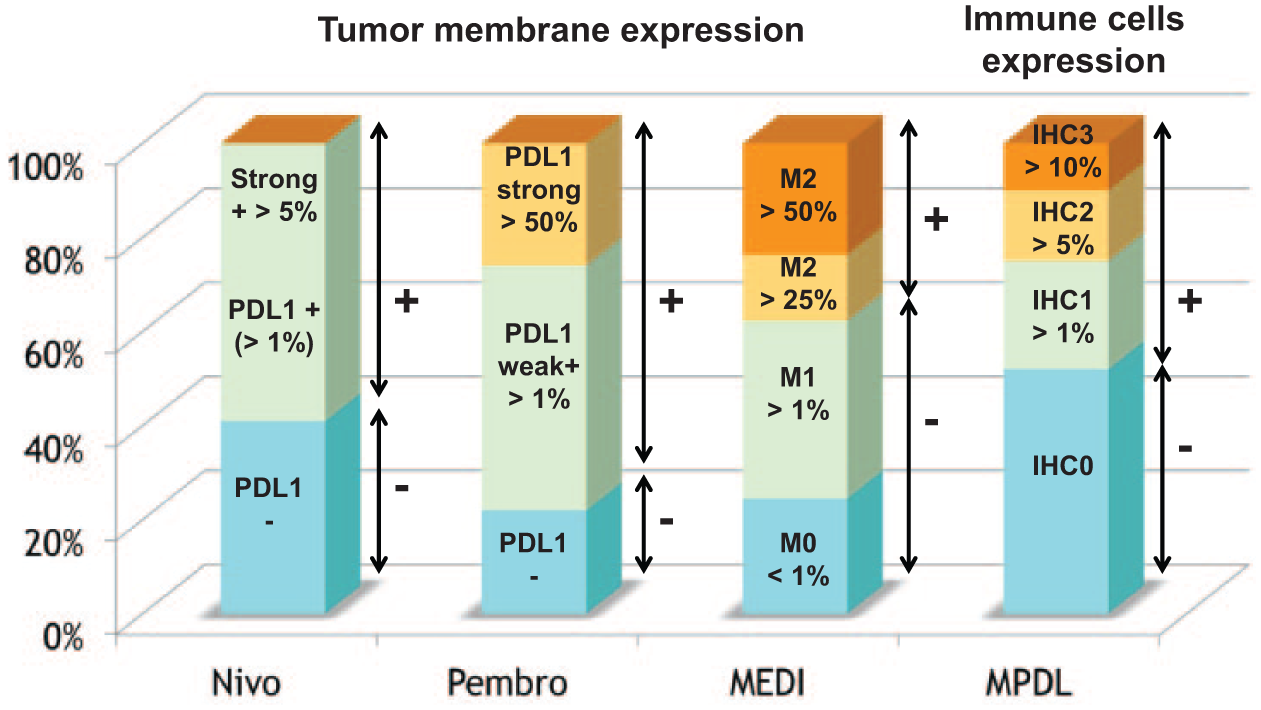
Strong arguments now exist to consider PDL1 expression in immunohistochemistry (IHC) analysis as a potential predictive biomarker for the response to drugs that target the PDL1/PD1 pathway. In the MPDL3280A phase I study, ORR was 46% (6/13) in patients with a PDL1 IHC score of 2 or 3, and 83% (5/6) in those with a PDL1 IHC score of 3 [Horn et al. 2013]. MEDI4736 response rate was also strongly correlated with PDL1 expression (39% versus 5% ORR) [Segal et al. 2014]. Response rates in the KEYNOTE-001 study (pembrolizumab) were 37%, 17%, and 10% for strong positive, weak positive, or negative PDL1 expression; respectively [Garon et al. 2014]. Our feeling is that PDL1 expression is not yet a perfect marker, as the optimal test has not been clearly defined and some patients who are PDL1– derive a benefit from these drugs even if the test is negative. Nevertheless, it appears to us to be an interesting basis for selecting patients, especially in the first-line setting, as chemotherapy (which can be associated with bevacizumab and maintenance therapy) is associated with a response rate and a PFS that can reach 35% and 7.4 months [Barlesi et al. 2013; Sandler et al. 2006], respectively. Moreover, recent studies underlined the potential predictive factor of immune markers not only in the tumor cells but also in the surrounding immune cells. Tumeh and colleagues showed that CD8, PD1, and PDL1 expression obtained from pretherapeutic biopsies in the tumor and at the invasive margin significantly correlated with response to pembrolizumab in melanoma [Tumeh et al. 2014]. Similarly, in other studies across multiple cancer types, responses to MPDL3280A were observed in patients with tumors expressing high levels of PDL1, especially when PDL1 was expressed by tumor-infiltrating immune cells. Furthermore, responses were associated with Th1 gene expression, CTLA4 expression, and the absence of fractalkine (CX3CL1) in baseline tumor specimens [Herbst et al. 2014]. We can thus anticipate that biomarkers might be dispensable in heavily pretreated patients who have no other therapeutic options, but will become mandatory in pretreated patients. The main characteristics of the PDL1 test used by different companies and the cutoff values are reported in Table 3 and Figure 3.
Biomarker test for PDL1 expression according to the companies.

IHC, immunohistochemistry; FFPE, formalin-fixed, paraffin-embedded.
Immunotherapy in the early stage of disease
Initial assessments of active immunotherapies have been conducted in patients with advanced-stage disease. However, rapid tumor growth and extensive tumor-related immunosuppression suggest that these patients might be least likely to benefit from this treatment. Another perspective in the field is thus to extend the indication of immunotherapy in the disease’s earlier stages. The 5-year survival for patients treated with surgery alone or with adjuvant chemotherapy remains low, underlining the need for new strategies. Moreover, not all patients with early-stage disease are eligible or willing to undergo chemotherapy following complete surgical resection. In the context of early-stage NSCLC, immunotherapeutic interventions that can both induce cell-mediated immunity against proliferating cancer cells following complete resection, and establish immunologic memory that may guard against future recurrences through active immune surveillance, are particularly attractive therapeutic options. Trials are about to begin in this setting (BR31-IFCT1401) [ClinicalTrials.gov identifier: NCT02273375]. Neoadjuvant trials are also interesting strategies that can initiate an immune response before surgery, even if the specific toxicity profile has to be addressed first in small phase I/II studies.
Conclusion
Enhancing the immune system in subjects with lung cancer represents an appealing therapeutic venue. As some immunotherapies have demonstrated significant activity in lung cancer, they may soon become a powerful addition to the oncologist’s toolbox. More basic research and clinical trials are needed to define which tumors (histology, size, stage), which patients (age, pretreatments, smoking habits), and which combinations (alone, with chemotherapies, or targeted therapies) are the best candidates.
Footnotes
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.