
Abstract
Background and Objectives
Trained immunity (TI) refers to a non-specific, long-lasting protective immune response that occurs following initial stimulation of the immune system and thought to be largely mediated by functional reprogramming of myeloid cells. TI has been demonstrated in BCG-vaccinated infants and can be induced in human cells and adult mice via agonists of pattern recognition receptors (PRR), such as β-glucan or MDP (a muropeptide that activates NOD2). However, its induction in neonates remains poorly understood. Previously, we demonstrated that the synthetic TLR2-NOD2 dual agonist CL429 enhances antimicrobial functions in adult mice and protects against subsequent infection with Leptospira interrogans, a zoonotic pathogen. We also demonstrated the immediate protective benefits of NOD2 stimulation in neonates against Cryptosporidium, a zoonotic pathogen that affects young animals in livestock herds. However, whether NOD2 agonists can induce TI in neonates and protect them at adulthood is unknown.
Methods and Results
Here, we investigated whether exposure of neonatal mice to PRR agonists (CL429, MDP or β-glucan), administered intraperitoneally at one week interval at 7/14 or 14/21 days of age, could enhance inflammatory cytokines production after ex vivo restimulation and confer long-term protection into adulthood. Surprisingly, none of the treatments enhanced ex vivo cytokine responses in adulthood after restimulation, nor did they confer protection against experimental leptospirosis. Instead, MDP-treated neonates exhibited 50% mortality following adult infection, revealing an unexpected detrimental effect.
Conclusion
These findings demonstrate that these PRR agonists fail to induce protective TI against Leptospira when administered to neonatal mice, challenging assumptions derived from adult models. Furthermore, our study reveals the risks of administering immunostimulants during the early stages of life, and highlights unanticipated and potentially harmful, PRR- and age-specific mechanisms of immune system modulation.
Introduction
The innate immune system serves as a first line of defense by detecting microbe-associated molecular patterns (MAMPs) as danger signals through pattern recognition receptors (PRRs), including Toll-like receptors (TLRs), NOD-like receptors (NLRs) and C-type lectin receptors (CLRs). Activation of these receptors triggers host defense mechanisms, with cytokines, chemokines and antimicrobial compounds production, leading to inflammation and pathogen clearance. 1 Emerging evidence suggests that PRR engagement can also induce long-lasting functional reprogramming of innate immune cells, a phenomenon termed “trained immunity” (TI). 2
First described in 2011, by Netea et al., TI refers to the epigenetic rewiring of innate immune cells (monocytes/macrophages, dendritic cells, neutrophils, and NK cells) that occurs following an initial exposure to pathogens or their products, leading to an enhanced non-specific response upon secondary challenge, 3 Unlike adaptive immunity, TI operates independently of T- and B- cells, offering broad-spectrum and long-term protection against unrelated infections. TI has been extensively studied in humans with documented benefits in infectious or cancer contexts,4–7 particularly following Bacillus Calmette-Guérin (BCG) vaccination, via the activation of NOD2, 8 the receptor of MDP, a muropeptide released by bacterial peptidoglycan. Notably, neonatal BCG vaccination confers protection against unrelated infections,9,10 supported by epigenetic DNA methylation in circulating monocytes from vaccinated infants. 11
Beyond BCG, fungal β-glucans recognized by Dectin-1 have been shown to trigger TI with evidence of long-term and broad protection against secondary infections. The mechanism involves epigenetic rewiring of hematopoietic stem cells, leading to enhanced bactericidal host defenses and cytokines production.3,12 Furthermore, in addition to being highly beneficial as vaccine adjuvants, several synthetic TLR agonists have also been shown to trigger trained immunity.13–17 TI can also be induced by stimulation of NOD2 receptor by synthetic MDP.18,19
TI has been shown in mice,3,20 inducing metabolic reprograming 21 and protection against infections with Listeria 22 or Mycobacterium tuberculosis. 12 Our group recently demonstrated that CL429, a dual TLR2/NOD2 agonist, induces TI in adult mice, improving control of Leptospira interrogans infection, 19 measured by live imaging of mice infected with bioluminescent leptospires. 23 The enhanced protection was associated to peritoneal, spleen and bone marrow macrophages that upon restimulation produced ex vivo more pro-inflammatory cytokines, and chemokines, as well as more nitric oxide (NO), a potent antibacterial compound. These effects were local, systemic, and long-lasting, underscoring the prophylactic potential of TI. 19
However, neonates are characterized by a distinct immunological profile, with functionally different innate and adaptive responses that affect their susceptibility to infection.24,25 For these individuals, strengthening their immune system appears to be a promising alternative for combating the severity of infections. Our group demonstrated that administration of TLR3, TLR9, or NOD2 agonists in neonatal mice can reduce cryptosporidiosis, a zoonotic disease affecting young animals,26–28 but neonatal TI remains poorly explored. Previous studies suggests its feasibility: TLR4 and TLR7/8 agonists mitigated sepsis in neonatal mice 29 and administration of TLR9 agonists to newborn broiler chicken protected them against experimental Escherichia coli septicemia at 27 days of age. 30 Yet, whether canonical TI-inducing agonists (CL429, MDP and β-glucan) can safely and effectively modulate neonatal immunity remains unknown.
To address this gap, we investigated whether intraperitoneal (IP) administration of MDP (NOD2 agonist), CL429 (TLR2/NOD2 agonist) and β-glucan (Dectin-1 agonist) could induce TI in murine neonates. We evaluated non-specific ex vivo cytokine responses and in vivo protection against Leptospira infection in adulthood. Surprisingly, none of the treatments conferred benefit; instead, MDP elicited adverse delayed effects, suggesting that these PRR agonists which have an established ability to induce TI in adults, may not translate safely or effectively in neonates. These findings emphasize the importance of exercising caution when immunostimulating neonates and highlight the need for age-specific TI strategies.
Material and methods
Animals - Ethics statements
Seven-to 21-day-old female and male C57BL/6J or albino C57BL/6 B6(Cg)-Tyrc-2J/J mice, and 7- to 12-week-old male and female C57BL/6J mice (bred in the animal facility at Institut Pasteur or the Infectiology of Farm, Model and Wildlife Animals Facility (PFIE) Centre INRAE Val de Loire (https://www6.val-de-loire.inrae.fr/pfie), or purchased from Janvier Labs, France) were used in this study. The mice were kept in individual ventilated cages with HEPA filters with food and water ad libitum. All experimental protocols were conducted in compliance with French legislation (Décret: 2001-464 29/05/01) and European regulations (86/609/CEE) governing the care and use of laboratory animals. This project was approved by the local ethics committee for animal experimentation: Comité d’Ethique pour l’Expérimentation Animale Val de Loire [CEEA VdL n°019: APAFIS#37335] and the Institut Pasteur ethics committee [Comité n°089: APAFIS#31815].
Leptospira interrogans cultures
Leptospira interrogans serovars Manilae strain L495, its bioluminescent derivative strain MFLum1, Copenhageni strain Fiocruz L1-130, and Icterohaemorrhagiae strain Verdun (Virulent Cl3) were grown in liquid Ellinghausen-McCullough-Johnson-Harris (EMJH) medium at 30°C without agitation, with weekly subculture. All the strains were used between passages 3 and 12, at the end of the log phase. Leptospires were centrifuged at 3,200 x g for 25 min at room temperature, resuspended in endotoxin-free D-PBS (Lonza) and enumerated using a Petroff-Hauser chamber. Inactivated heat-killed (HK) leptospires were heated at 56°C for 30 min with mild agitation.
Mouse treatments
The PRR agonists MDP (#tlrl-mdp, InvivoGen), CL429 VacciGrade™ (#vac-c429, InvivoGen), and β-glucan (#G5011, Sigma) were used as immunostimulants to promote the establishment of trained immunity in neonates and adults C57BL/6J or albino C57BL/6 B6(Cg)-Tyrc-2J/J mice. Each experimental group was a mix of mice from different litters to avoid a biased effect. In neonates, the immunostimulants were administered intraperitoneally on days 7 and 14, or days 14 and 21 after birth. For adults, the first administration occurred between 6 and 8 weeks of age, followed by a second injection one week later. Neonatal mice, either 7-14 days or 14-21 days old at the time of injection, were referred to as early or late neonates, respectively. They received 5 µg of CL429, 200 µg of β-glucan, or 20 µg of MDP in a final volume of 20 µL. Adult mice were injected with 1 mg β-glucan (in 200µL) as described. 12 Control mice were injected with the corresponding vehicle (usually D-PBS, referred to as PBS, or 5% DMSO-PBS when CL429 was used). To confirm the basal immunostimulatory effects of PRR agonists in neonatal mice, CL429, β-glucan, or MDP were administered intraperitoneally at the aforementioned doses to eight-day-old neonatal mice. Non-treated controls received an equivalent volume of PBS, or DMSO-PBS. The mice were sacrificed 4 hours post-injection. Whole blood was collected, and sera were stored at −20°C for cytokine analysis.
Harvest and ex vivo stimulation of peritoneal cells
One or three weeks after the second administration of immunostimulants, the mice were euthanized, and the peritoneal cells were collected to assess the immune inflammatory response. Peritoneal lavage was performed by injecting into the peritoneal cavity 3 mL of complete cold RPMI (RPMI GlutaMax (Gibco), 10% heat-decomplemented Foetal calf serum (FCS) (Lonza), 1 mM HEPES (Gibco), 1 mM non-essential amino acids (Gibco), and 1 mM sodium pyruvate (Gibco)) supplemented with 1X antibiotic-antimycotic (Anti-Anti Gibco). The peritoneal content was passed through a 100 μm strainer and centrifuged. Only pellets free of red blood cell contamination were used. Peritoneal cells (2×105 cells/well) were seeded into a 96-well plate (TPP). After 2-3 h of incubation at 37°C and 5% CO2, the cells were washed once with D-PBS to remove the non-adherent cells. The peritoneal cells, mostly macrophages, were stimulated for 24 h with 100 ng/mL E. coli LPS (EB InvivoGen), heat-killed Escherichia coli (K12J5 RifR, 30 min at 100°C, kindly provided by the PGBA INRAE team), heat-killed Bacille de Calmette et Guérin (BCG, 30 min at 120°C, kindly provided by Dr. Lagranderie and Dr. Marshall, Institut Pasteur), or with live or heat-killed L. interrogans (Manilae L495, Fiocruz L1-130, or Verdun) at a multiplicity of infection (MOI) of 100.
In vivo infection and live imaging (IVIS)
The long-term protective effect against Leptospira infection was evaluated between 4 to 6 weeks after the administration of PRR agonists to neonates. Mice (both male and female) were infected intraperitoneally with 2×107 leptospires/mouse, which corresponds to a sublethal dose, in a final volume of 200 μL of PBS. The pathogenic Manilae bioluminescent MFlum1 strain was used to assess the course of the infection overtime using live imaging (IVIS Imaging system). The control groups consisted of uninfected mice that were treated with agonists as neonates, or mice that were injected with vehicle as neonates, and infected in adulthood.
After infection, a clinical score was determined based on general and specific parameters, such as body weight changes and clinical signs, as previously described. 31 Images monitoring the bacterial dissemination during the acute phase or renal colonization were acquired using an IVIS Spectrum system (Xenogen Corp., Alameda, CA) according to instructions from the manufacturer, as previously described. 32 Quantitative data for the average light flux normalized by time, and the area of regions of interest (ROIs), were defined as light units per second, per cm2, per steradian (p/s/cm2/sr). Images were analyzed using Living Image software (Perkin Elmer), with the photon count adjusted to 1×104 to 1×106 (p/s/cm2/sr) for all images shown. Mice were euthanized by CO2 exposure or cervical dislocation 1-month post-infection.
Nitric oxide and cytokine quantification
Nitric oxide (NO) was quantified using the Griess reaction in 50 µL of the cell supernatant 24 hours after ex vivo stimulation and immediately after collection. The cell supernatants were stored at -20˚C until cytokine dosages. Interleukin (IL)-6, IL-1β, CCL5 and KC levels were quantified by ELISA using a mouse R&D Systems DuoSet ELISA Kit, according to the manufacturer’s instructions. Optical density was measured at 450 nm, and cytokine concentration was determined using a standard curve.
Statistics
Statistical analyses were performed using the GraphPad Prism v10 software (GraphPad Software). Given the relatively small sample sizes (n = 6-8 per group), normality could not be reliably assessed; therefore, a nonparametric Mann-Whitney test was applied to evaluate differences between two independent groups. P values of less than 0.05 were considered statistically significant. p values: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
CL429 treatment in early neonatal mice does not induce enhancement of local trained immunity
A previous study has shown that the dual agonist CL429, stimulating NOD2 and TLR2, is able to provide trained immune responses in adult mice and protect them against a subsequent Leptospira interrogans infection. The mechanism was found to be associated with enhanced bactericidal functions of macrophages upon restimulation, and with increased production of the proinflammatory cytokines IL-6 and IL-1β, as well as the RANTES/CCL-5 chemokine,
19
a marker of Leptospira infection.
33
We investigated whether the process of trained immunity can also be induced in the neonatal period. Before investigating trained immunity, we first tested the compound’s ability to stimulate the neonatal innate immune system. We intraperitoneally (IP) injected eight-day-old mice with CL429 (5µg) and measured the production of inflammatory cytokines in blood 4 hours later. As shown in Figure 1(a), IL-6 levels in mice sera increased after CL429 treatment compared to the non-treated mice (PBS). However, no secretion of IL-1β or CCL5 was observed. Then, we investigated whether CL429 could induce a trained immune response. As previously described in adults,
19
neonatal mice received two IP injections, one week apart, of CL429 (5µg) at 7 and 14 days after birth. One week later, the intraperitoneal cells were collected to measure the inflammatory response ex vivo after restimulation (Figure 1(b)). IP cells were stimulated for 24 hours with Escherichia coli (E. coli) LPS or with different heat-killed bacteria (HK): Leptospira interrogans (L. interrogans; L. i) serovars Manilae strain L495, Copenhageni strain Fiocruz L1-130, and Icterohaemorrhagiae strain Verdun, E. coli, or Bacillus Calmette-Guérin (BCG) (Figure 1(b)). We showed that CL429 treatment did not enhance inflammatory responses compared to the PBS control group. Indeed, IL-6, IL-1β, and CCL5 levels were not modified after restimulation with LPS or HK bacteria (Figure 1(b)), regardless the type of bacteria. CL429 treatment in early neonates does not enhance inflammatory responses after ex vivo restimulation. (a) Chronogram of treatment of 8-day-old mice intraperitoneally (IP) injected with CL429 (5 μg) or PBS and sacrificed 4 h later (hours + 4). Graphs represent the production of cytokines in serum determined by ELISA. Data are from 2 independent experiments and pooled with n = 5 or 6 mice per group. (b) Chronogram of CL429 treatment of neonatal mice IP injected on day 7 and 14 after birth with PBS (n = 6) or 5 µg CL429 (n = 7), and assessment of the ex vivo responses of the peritoneal cells one week later (week + 1). Cells were stimulated ex vivo with 100 ng/mL LPS (E. coli) or heat-killed bacteria: Leptospira interrogans serovar Manilae strain L495, serovar Copenhageni strain Fiocruz L1-130 or serovar Icterohaemorrhagiae strain Verdun and Escherichia coli (E. coli) or Bacille de Calmette et Guérin (BCG) at a multiplicity of infection (MOI) of 100. The graphs represent the production of cytokines as determined by ELISA in the supernatants of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS). The data are representative of 3 independent experiments. Each point corresponds to one mouse and bars represent the median. Mann-Whitney test; ns = p-value > 0.05, ** p-value < 0.01.
β-glucan does not induce trained immunity in early neonates
Since CL429 was unable to induce trained immunity in neonates, we evaluated the effect of β-glucan, a fungal cell wall component, well known for providing trained immunity and protection against various infections in different mouse models.12,34–36 First, we assessed the inflammatory responses after β-glucan treatment in 8-day-old neonates. We administered a high dose (200 µg) of β-glucan, equivalent to the dose given to adults,
12
and measured the inflammatory response in the blood of the neonates 4 hours after treatment. As observed with CL429 (Figure 1(a)), we found that β-glucan injection induced IL-6 secretion, but not IL-1β or CCL5 (Figure 2(a)). Next, we injected neonatal mice twice, on days 7 and 14, to determine whether β-glucan could enhance the immune responses in neonates’ IP cells one week after the second treatment (Figure 2(b)). IP cells were stimulated with LPS (E. coli) or with HK bacteria: L. i (serovars Manilae strain L495, Copenhageni strain Fiocruz L1-130, and Icterohaemorrhagiae strain Verdun), E. coli, and BCG. Unexpectedly, treatment with β-glucan in neonates did not enhance the inflammatory immune response compared to the PBS control group (Figure 2(b)). Indeed, the levels of IL-6, IL-1β, and CCL5 were not increased after restimulation with LPS or HK bacteria. Furthermore, we noted that IL-6 responses in β-glucan-treated neonates were lower than in the PBS group and were significantly reduced after restimulation with HK L. i Fiocruz and Verdun strains. The same was observed for IL-1β responses following restimulation with HK E. coli and the L. i Verdun strain. Overall, we observed an overall trend of decreased inflammation after cell restimulation following β-glucan treatment of neonates. β-glucan, the iconic inducer of trained immunity, fails to enhance innate responses in early neonates upon ex vivo restimulation. (a) Chronogram of treatment in 8-day-old neonatal mice injected with β-glucan (200 µg) or PBS and sacrificed 4 hours later (hours + 4) for blood collection. Graphs show cytokines production in serum determined by ELISA. Data correspond to 2 independent experiments treated individually and pooled with n = 5 or n = 6 mice per group. (b) Chronogram of treatment in neonatal mice IP injected on day 7 and 14 after birth with 200 µg β-glucan (n = 6) or PBS (n = 6), and assessment of the ex vivo response in peritoneal cells harvested one week later (week + 1). Peritoneal cells were stimulated ex vivo with 100 ng/mL LPS (E. coli) or heat-killed bacteria: Leptospira interrogans serovar Manilae strain L495, serovar Copenhageni strain Fiocruz L1-130 or serovar Icterohaemorrhagiae strain Verdun and Escherichia coli (E. coli) or Bacille de Calmette et Guérin (BCG) at a multiplicity of infection (MOI) of 100. Graphs show cytokines production determined by ELISA in the supernatant of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS). The data are representative of 3 independent experiments. Each point corresponds to one mouse and bars represent the median. Mann-Whitney test; ns = p-value > 0.05, * p-value < 0.05, ** p-value < 0.01.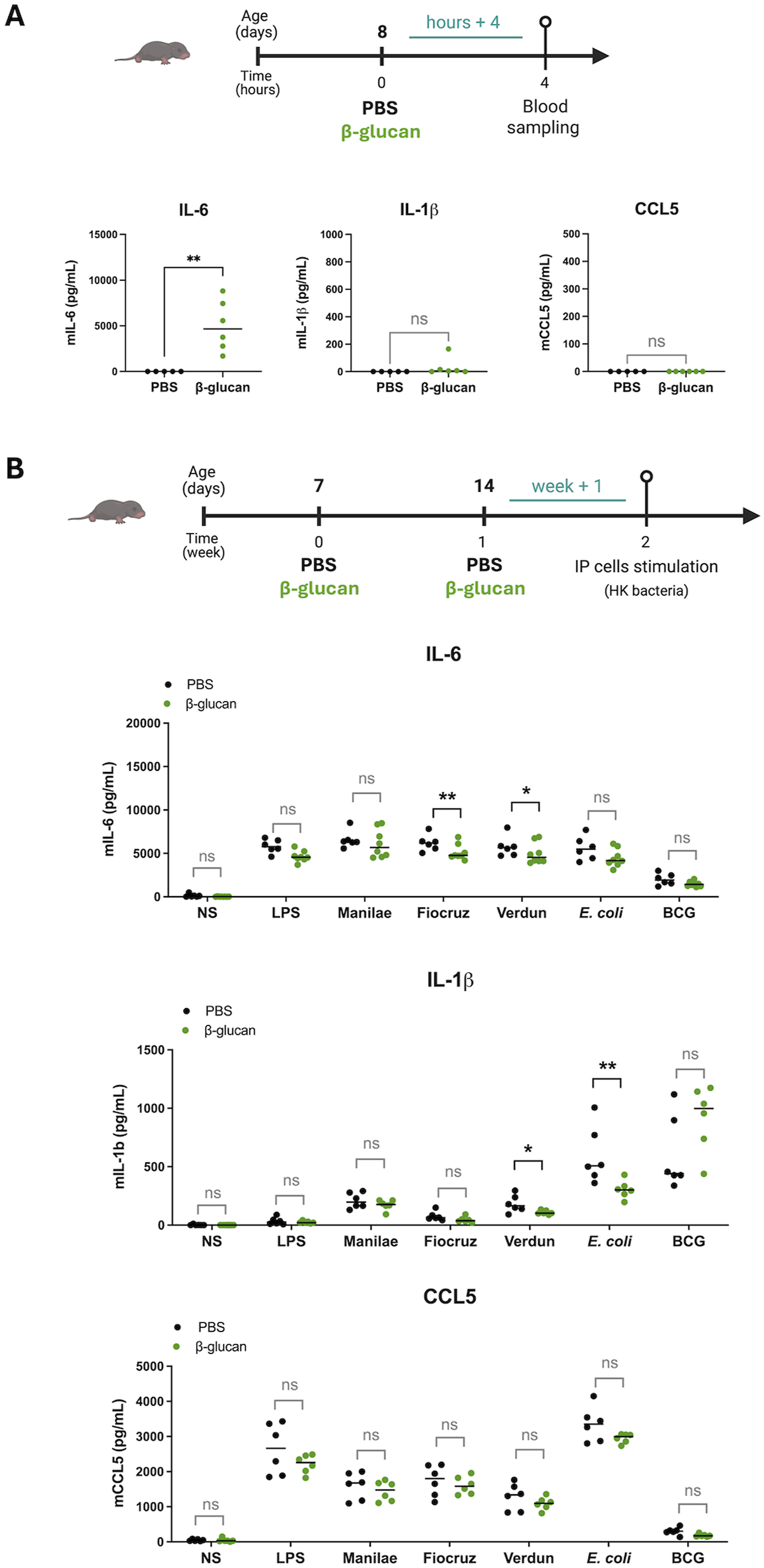
To control our experiment in neonates, we studied the effect of β-glucan, which is known to induce trained immunity in adult mice, under the same settings. 12 Adult mice aged 6–8 weeks were injected twice with 1mg of β-glucan, one week apart. One week later, we measured the innate immune responses ex vivo after restimulation (Sup Figure 1). IP cells were collected and stimulated with LPS (E. coli) and HK bacteria, as above. As expected, we demonstrated that, unlike IP cells from neonates, cells from β-glucan-treated adults exhibited a significant increase in IL-6, IL-1β and CCL5 levels to most stimuli compared to the control PBS group (Sup Figure 1), showing that the β-glucan was active and indeed induced TI in adult mice, although it did not induce TI in early neonates.
Delayed administration of CL429 or β-glucan in late neonatal mice does not enhance innate immune inflammatory responses
Since immunostimulant treatments (CL429 and β-glucan) that were effective in adults (
19
and Sup Figure 1) did not enhance inflammatory responses in neonates IP cells when administered at days 7 and 14 (Figures 1 and 2), we questioned whether the timing of administration of these immunostimulants was appropriate. To target the innate immune system at a more mature developmental stage, we changed the treatment protocol and administered the CL429 and β-glucan immunostimulants in late neonates on days 14 and 21 (Figure 3). One week after the last treatment, IP cells were collected and stimulated ex vivo with LPS (E. coli) and HK bacteria, as before. Our results showed that delaying the immunostimulation did not enhance inflammatory responses following the ex vivo restimulation of IP cells. In fact, IL-6, IL-1β and CCL5 levels were not increased in late neonatal mice treated with CL429 or β-glucan compared to the PBS control group (Figure 3). Furthermore, as before, we observed lower overall levels of cytokine production in animals treated with β-glucan than in the PBS control group. This was significant after LPS and L. i. Manilae, Fiocruz, and Verdun stimulations for IL-6 responses (Figure 3). There were also significantly decreased CCL5 responses in late neonatal mice treated with β-glucan after LPS and L. i. Manilae stimulations, as well as in CL429-treated late neonatal mice after E. coli and BCG stimulations (Figure 3). Delayed administrations of CL429 or β-glucan to late neonatal mice do not overcome failure to induce trained immune response. Chronogram of treatment to young mice IP injected on day 14 and 21 after birth with 5 μg CL429 (n = 6), or 200 µg β-glucan (n = 6), or PBS (n = 6), and assessment of ex vivo response one week later (week + 1). Peritoneal cells were stimulated ex vivo with 100 ng/mL LPS (E. coli) or heat-killed bacteria: Leptospira interrogans serovar Manilae strain L495, serovar Copenhageni strain Fiocruz L1-130 or serovar Icterohaemorrhagiae strain Verdun and Escherichia coli (E. coli) or Bacille de Calmette et Guérin (BCG) at a multiplicity of infection (MOI) of 100. The graphs represent the cytokine production as determined by ELISA in the supernatant of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS), depending on the agonist treatment (CL429 or β-glucan). The data are representative of 2 independent experiments. Each point corresponds to one mouse and bars represent the median. Mann-Whitney test; ns = p-value > 0.05, * p-value < 0.05, ** p-value < 0.01.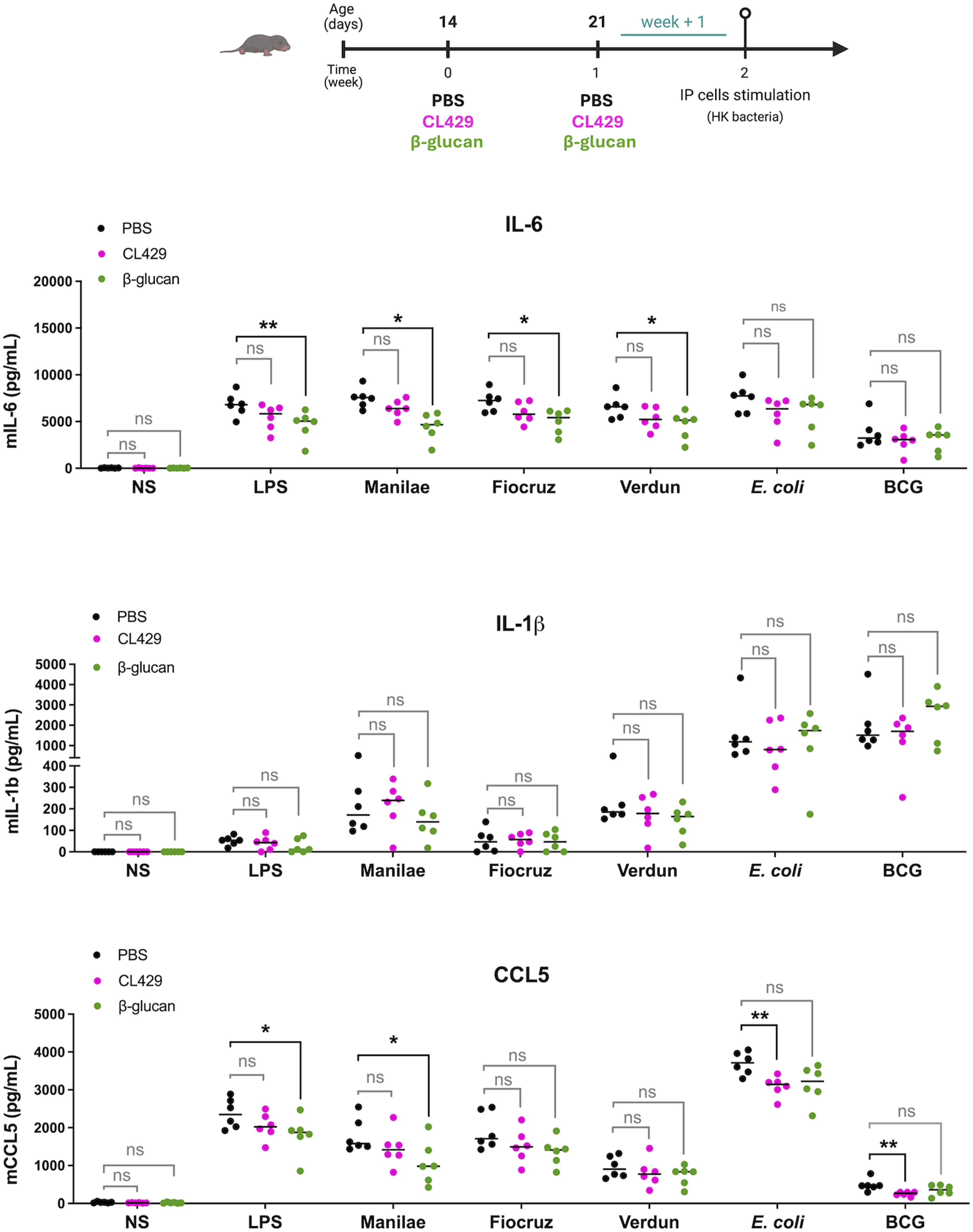
Immune responses to live L. interrogans infection remain unchanged after CL429 or β-glucan treatment in late neonatal mice
Considering these results, we wondered whether injecting these immunostimulants during the neonatal period might elicit better or worse responses to Leptospira infection once mice reached adulthood. The first step towards achieving this goal was shifting the restimulation window for IP cells to three weeks (week +3) after the final immunostimulant injection (PBS, CL429, or β-glucan) administered on days 14 and 21 (Figure 4(a)), to mimic the state of the mice at the time of a potential adult infection. At six weeks of age, the mice were sacrificed, and the IP cells were collected and stimulated ex vivo with LPS (E. coli), or instead of HK bacteria as before, with live leptospires (L. interrogans serovars Manilae strain L495, Copenhageni strain Fiocruz L1-130, and Icterohaemorrhagiae strain Verdun). First, nitric oxide (NO) assays were performed to measure local inflammation. Regardless of the immunostimulant used, we found no difference in NO production in CL429-or β-glucan-treated animals compared to the PBS control group (Figure 5(a)). We then measured inflammatory cytokine responses which showed that, like NO responses, the levels of IL-6, IL-1β and CCL5 cytokines were not increased in mice treated with CL429 (Figure 5(b), upper panels) or β-glucan (Figure 5(b), bottom panels), compared to the PBS control group. In general, we observed a trend to lower responses in mice treated with β-glucan than in the PBS group. Notably, there was a significant decrease in the IL-6 response after L. i Manilae stimulation in β-glucan treated mice (Figure 5(b), bottom panel). CL429 and β-glucan treatments in late neonatal mice do not induce trained immune responses after ex vivo restimulation with live L. interrogans. (a) Chronogram of treatment: Mice were IP injected with 5 μg CL429, or 200 µg β-glucan, or PBS on day 14 and 21 after birth and sacrificed 3 weeks later (week + 3). Peritoneal cells were stimulated ex vivo with 100 ng/mL LPS (E. coli) or live Leptospira interrogans serovar Manilae strain L495, serovar Copenhageni strain Fiocruz L1-130, or serovar Icterohaemorrhagiae strain Verdun, at a multiplicity of infection (MOI) of 100. The graphs represent the production of nitric oxide (NO) measured by the Griess method in supernatant of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS), depending on CL429 (left panel) or β-glucan (right panel) treatment. (b) Graphs represent the secretion of cytokines determined by ELISA in the supernatant of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS), depending on CL429 (upper panels) or β-glucan treatment (bottom panels). Data are representative of 3 independent experiments. Each point corresponds to one mouse and bars represent the median, n = 4 to 8 mice per group. Mann-Whitney test; ns = p-value > 0.05, * p-value < 0.05. MDP injections in late neonatal mice do not enhance cytokine responses after ex vivo restimulation. (a) Chronogram of MDP treatment in neonatal mice and serum collection. Eight-day-old mice were IP injected with PBS or 20 µg MDP and sacrificed 4 h later (hours + 4) to collect blood. Graphs represent the production of cytokines in serum determined by ELISA. Data are from 2 independent experiments and pooled with n = 5 or 6 mice per group. (b) Chronogram of treatment: Mice were IP injected with PBS (n=5) or 20 µg MDP (n = 5) on day 14 and 21 after birth and sacrificed 3 weeks later (week + 3). Peritoneal cells were stimulated ex vivo with 100 ng/mL LPS (E. coli) or live Leptospira interrogans (L.i) serovar Manilae strain L495 at a MOI of 100. Upper panel represents the production of nitric oxide (NO) measured by Griess method in the supernatant of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS). Graphs from the lower panels show the secretion of cytokines determined by ELISA in the supernatant of peritoneal cells 24 h post-stimulation versus non-stimulated cells (NS). Data are representative of 3 independent experiments. Each point corresponds to one mouse and bars represent the median. Mann-Whitney test; ns = p-value > 0.05, ** p-value < 0.01.
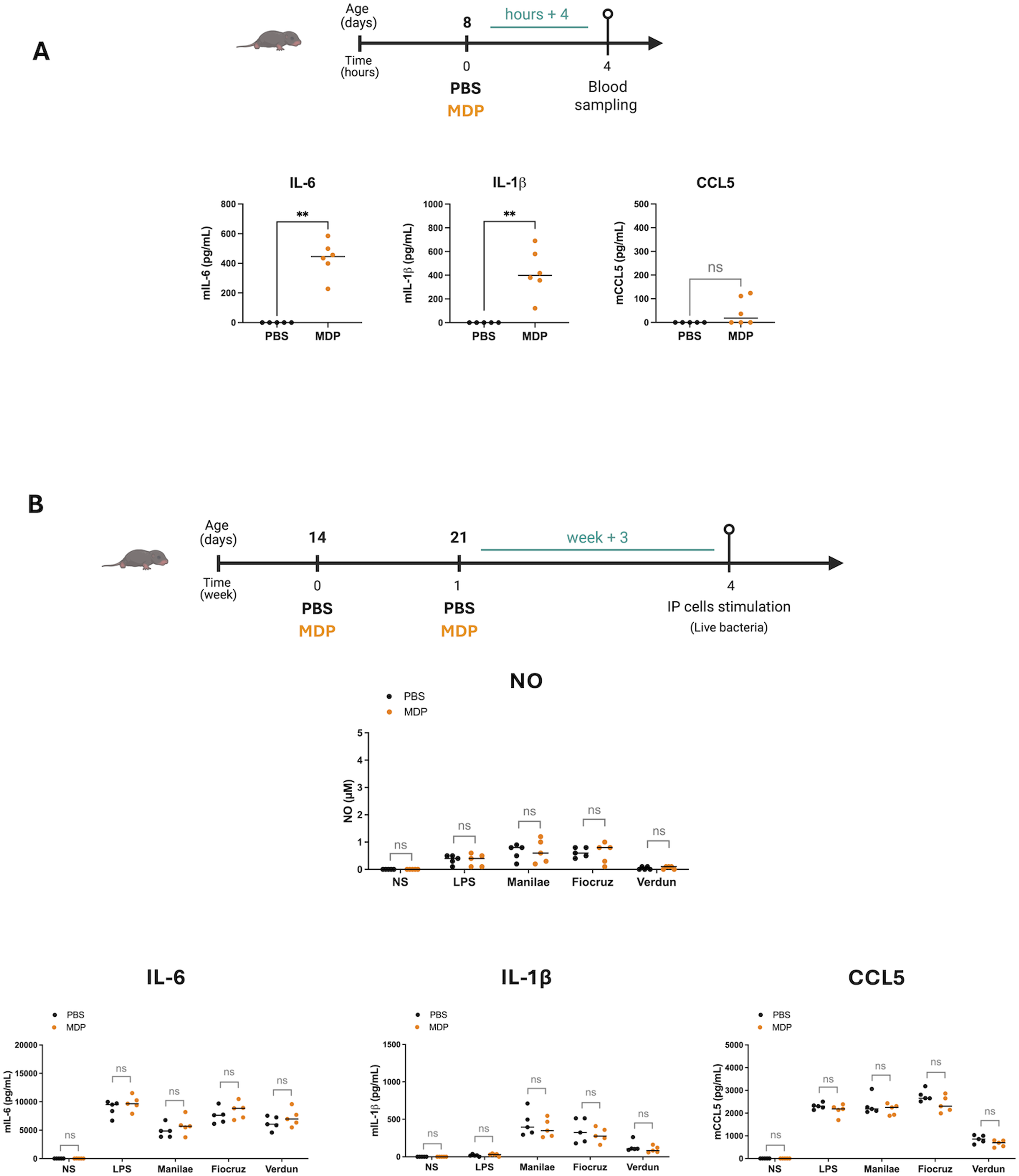
MDP, the NOD2 ligand, administered in late neonatal mice doesn’t induce trained immunity.
Since both CL429 and β-glucan are complex molecules using different receptors, we evaluated the effect of MDP, the agonist of NOD2 receptor which is known to induce TI37,38 and to stimulate neonatal mice in an infectious context. 28 We intraperitoneally (IP) injected eight-day-old mice with MDP (20µg) and measured the production of inflammatory cytokines in the blood 4 hours later. IL-6 but also IL-1β levels in mice serum increased after MDP treatment compared to the non-treated mice (PBS). A trend but no significant increase in secretion CCL5 was observed (Figure 5(a)). We then investigated whether MDP could induce a trained immune response. Neonatal mice received two IP injections, one week apart, of MDP (20µg) at 14 and 21 days after birth. Three weeks later, the intraperitoneal cells were collected to measure the inflammatory response ex vivo after restimulation (Figure 5(b)). IP cells were stimulated for 24 hours ex vivo with LPS (E. coli) or with live leptospires (L. interrogans serovars Manilae strain L495, Copenhageni strain Fiocruz L1-130, and Icterohaemorrhagiae strain Verdun). Nitric oxide (NO) assays were performed to measure inflammation. We found no difference in NO production after MDP-treated animals compared to the PBS control group (Figure 5(b)). We then measured inflammatory cytokine responses which showed that, like NO responses, the levels of IL-6, IL-1β and CCL5 cytokines were not increased in mice treated with MDP-injection (Figure 5(b)) compared to the PBS control group regardless the strains of bacteria.
In vivo model of leptospirosis at adulthood reveals detrimental effects of early-life MDP treatment
After examining the above in vivo–ex vivo approach, we aimed to test whether early-life MDP treatment affects immune defense against adult infections. To address this question, neonatal albino (C57BL/6 B6(Cg)-Tyrc-2J/J) mice received as before two intraperitoneal injections of MDP (20 µg), administered one week apart, at 14 and 21 days after birth. Three weeks following the second injection, the animals were IP infected with the bioluminescent L. interrogans MFLum1 strain, a derivative of the L. interrogans serovar Manilae L495 strain 23 and 32, and the infection was tracked by live imaging at D0, D3, D7, D15 and D30 after infection (Figure 6(a)). No detectable differences in bacterial loads between MDP-treated mice and the PBS control group were observed (Figure 6(b)). However, mice that had received MDP during early life were unexpectedly more sensitive to leptospiral infection and some mice died from the infection, suggesting an altered and detrimental response to Leptospira. A second experiment confirmed that lethality was observed in the MDP-treated groups (Figure 6(c)). This lethality reached 50% of the total cohort and was not sex-dependent, as both males and females were equally affected (Figure 6(b)–(c)). We also observed that deaths occurred between days 4 and 7 post-infection. This timeframe coincides with the end of the acute phase, which corresponds to the septicemic phase and systemic dissemination of the bacteria. The bacterial load typically peaks at day 3 post-infection and gradually declines, becoming undetectable by days 6 to 7 post-infection in mice, indicating apparent clearance of Leptospira from the bloodstream.
23
This suggests that the MDP pretreated mice were unable to control the infection. The same experiment was conducted with β-glucan (Figure 7). As for the MDP-treated mice, no difference in bacterial loads between the β-glucan and PBS control groups was observed. However, in contrast to MDP treatment and despite the adverse effects previously detected in ex vivo assays (Figures 2–4), no lethality was observed in mice that had received early-life β-glucan treatment (Figure 7). A similar absence of in vivo detrimental outcomes was also seen in neonatal mice treated with CL429 (Sup Figure 2). Early-life immunostimulation by MDP shapes adult susceptibility and lethality to Leptospira. (a) Chronogram of treatment: albino (C57BL/6 B6(Cg)-Tyrc-2J/J) mice were IP injected with PBS or 20 µg MDP on days 14 and 21 after birth, and IP infected three weeks later (week +3) with 2x107 bacteria of the MFlum1 strain of L. interrogans, as previously described.
23
Mice were monitored by live imaging on days 0, 3, 7, 15 and 30 post-infection. All bioluminescence analyses were carried out after IP administration of D-luciferin. Two independent experiments (EXP) were conducted: EXP1 in (b), and the second experiment EXP2 in (c). Graph in left B panel shows the quantification of relative light units (RLU) expressed as mean ± SEM of average radiance of light measured in photons/second/cm2 in mice imaged from ventral (days 0 to 7) or dorsal (days 15 and 30) views. (b)–(c) Images show tracking of mice at different times post-infection. The blue to red scale is proportional to the intensity of bioluminescence, reflecting the number of live leptospires. Crosses indicate mice that died or were sacrificed for endpoint due to acute leptospirosis. (d) Table of lethality. The left table summarizes the number of mice included, the number of recorded deaths and the mortality rate by sex in each group of mice pretreated with (MDP) or PBS and infected with leptospires. The right panel shows the survival curve according to the groups of mice. Data correspond to 2 independent experiments, pooled with n=3 mice for the PBS control group and n=10 mice for the MDP-treated group. Early-life β-glucan treatment does not affect adult susceptibility to leptospirosis and mice survival. Chronogram of treatment: albino (C57BL/6 B6(Cg)-Tyrc-2J/J) mice were IP injected with PBS or 200 µg β-glucan on days 14 and 21 after birth, and IP infected three weeks later (week +3) with 2x107 bacteria of the MFlum1 strain of L. interrogans, as described previously.
23
Mice were monitored by live imaging on days 0, 3, 7, 15 and 30 post-infection. All bioluminescence analyses were carried out after IP administration of D-luciferin. The graph on the left shows the quantification of relative light units (RLU) expressed as mean ± SEM of average radiance of light measured in photons/second/cm2 in mice imaged from ventral or dorsal views. Images show tracking of mice at different times post-infection. The blue to red scale is proportional to the intensity of bioluminescence, reflecting the number of live leptospires.

Discussion
In this study, we tested the possibility to harness neonatal mice immune system to protect them at adulthood against infection. We used two different windows of treatments with CL429, β-glucan and MDP, three PRR agonists known to induce trained immunity (TI) in adult mice, to treat early and late neonates. However, no beneficial effect was observed, neither in an in vivo-ex vivo experiment with stimulation of peritoneal cells of mice previously treated with the agonist, nor when treated mice were infected at adulthood with Leptospira (Figure 8). Unexpectedly, MDP treatment of late neonates induced increased sensitivity to the infection, with observed lethality in mice, which never happened in control groups at this infecting dose.
23
This was even more unexpected since we previously demonstrated in neonate mice a therapeutic effect of MDP used at day 8 of age to alleviate experimental infection with Cryptosporidium parvum inoculated at day 3 of age.
28
Deleterious effects were also observed in peritoneal cells from mice previously treated with β-glucan, where decreased secretion of cytokines was observed, although β-glucan treatment in neonates did not cause death in Leptospira-infected adult mice.
These negative effects of trained immunity in neonates (here with β-glucan ex vivo and MDP in vivo) are reminiscent of our recent observation of the adverse effects of TLR2/TLR6 agonists causing death in neonates. 39 Likewise, it was shown that BCG vaccination aggravates necrotizing colitis in neonates. 40 Furthermore, it has been shown that IL-1β signalling played a key role in the induction of TI by β-glucan in adult mice and the protection against pulmonary infection due to Mycobacterium tuberculosis. 12 Of interest, in our study, only MDP induced IL1-β in sera after injection in neonates. We may hypothesize that in the case of neonates, IL1-β signalling could be at the origin of the deleterious imprinting.
This age-dependent adverse outcome suggests that neonatal immune programming by PRR agonists is context- and timing-dependent, with potential for paradoxical immunosuppression rather than enhancement.
The inability to induce TI could potentially be attributed in the difference in microbiota status between neonates and adults. Indeed, our experiments were performed during the suckling time (7-14 days old) and during the progressive establishment of weaning (14-21 days old), where the colonizing microbiota has a big influence on the immune response. 41 Indeed, the microbiota of suckling mice progressively diversifies until weaning, becoming more stable in adulthood. We may hypothesize that the age-related changes observed in species diversity,42,43 known to shape long-term host-microbial mutualism may also modulate the development of host defence and NOD1/2 and PRR responses, as it has been shown for proteobacteria (Gram negative bacteria) playing a crucial role in priming neutrophil functions through release of peptidoglycan fragments activating NOD1. 44 However, the minimal responses to the PPR agonists in the blood of eight-day-old newborns, and the lack of TI induction regardless of the time window for treatment administration, remain mechanistically unexplained. Considering human data, some hypotheses can be put forward: neonatal innate immune cells may have higher activation thresholds for PRRs due to blunted PRR expression. For example, TLR4 expression is lower in neonatal monocytes than in adult ones, and the expression of the TLR adaptor MyD88 is reduced. 45 The paucity of responses in mouse neonates may also be explained by altered downstream signalling, potentially favouring anti-inflammatory over pro-inflammatory cytokines, or having a higher baseline expression of regulators that dampen TLR signalling, as it has been described for SOCS1, 46 and hypothetically could also be the case for other PRR inhibitors, such as IRAK-M or A20.
Although in vivo induction of TI is well documented with BCG via NOD2 stimulation37,38,47 and β-glucan,12,35,48,49 our findings that administration of NOD2 and Dectin-1 agonists is not always beneficial in neonatal mice suggest that the complexity of agonists and/or persistence of activation may be important factors to induce TI. Indeed, BCG, a live attenuated M. tuberculosis vaccine, engages multiple PRRs, including TLRs, CLRs, NLRs, as well as NOD2, Dectin-1, and the mannose receptor.50,51 β-glucan is primarily recognized by Dectin-1, but its signalling also involves CR3, TLR2, and CD5.49,50 CL429, bi-specific ligand is recognized by TLR2/6 and the NOD2 receptor, 13 while MDP is recognized only by NOD2. 52 Of note, attenuated live bacteria like BCG provide continuous stimulation, whereas single ligands such as β-glucan, CL429, or MDP may induce more transient responses, raising questions about the duration and intensity of stimulation. Supporting this, TI is more effectively induced when BCG treatment is supplemented with bacterial lipoproteins (BLPs) compared with BCG or BLPs alone. 47
The mechanisms underlying TI are increasingly described, mainly in vitro,3,8,53 but in vivo validation remains limited.19,48 Unlike in vitro studies with defined stimulation-resting schedules, in vivo studies of TI lack standardized protocols and show significant variability in methodology, which makes comparisons difficult. This diversity of approaches raises questions about the choice of inducer, induction strategy, timing of analysis, and choice of readouts. Indeed, we may also acknowledge the possibility that neonatal TI may manifest through alternative outputs (e.g., antimicrobial activity, phagocytosis, survival signalling) rather than classical inflammatory cytokines.
Our group focuses on immune functions, particularly innate immunity, during the neonatal period. Our choice was to use two complementary approaches: the in vivo–ex vivo model, where cells are purified after in vivo stimulation and restimulated ex vivo, and the in vivo–in vivo model, where animals receive a second in vivo challenge at the time studied before ex vivo, allowing for better understanding of the effect of treatments. In our study, we aimed to obtain TI through two injections of immunostimulants, according to our previous study in adult mice using CL429. 19 Literature reports variability, with one or two inductions effective depending on the ligand and study.47–49 For example, a single β-glucan injection protects against Leishmania braziliensis. 35 Two β-glucan injections are more common, effectively protecting against Pseudomonas aeruginosa, 49 Mycobacterium tuberculosis, 12 or LPS rechallenge, 48 with interval between injections varying from 2 to 4 days.48,49 Overall, induction frequency varies, yet each model effectively induces trained immunity.
In addition, most in vivo studies analyzed TI within 1 to 7 days after the last injection.19,35,48 Protective effects are typically transient, declining beyond 7 days,35,48 suggesting that ligands like β-glucan do not induce long-term memory without repeated stimulation. In our neonatal model, β-glucan administration reduced cytokine responses of peritoneal cells ex vivo, and similar effects were observed with MDP in the in vivo–in vivo model, suggesting that neonatal immune stimulation may induce tolerance or long-term hypo-responsiveness rather than trained immunity. However, we can conclude that the agonists did not trigger immune paralysis since the peritoneal cells from treated neonates responded as the non-treated one.
Trained immunity and innate immune tolerance are opposite outcomes of innate immune memory: the former enhances effector responses upon restimulation, while the latter suppresses them.54,55 Both rely on epigenetic and metabolic reprogramming but produce distinct functional outcomes depending on the initial stimulus. 54 Epigenetic changes are generally impaired in neonates due to the reduced activity of epigenetic writers or a closed chromatin state at the promoters of key inflammatory genes.56,57 Zhou et al. demonstrated that treating neonatal mice with BCG plus bacterial lipoprotein induces protective trained immunity against microbial sepsis. 47 This work is important because, in contrast to our single TLR agonist protocol, stimulation based on complex agonists resulted in histone modifications with enhanced H3K4me3 and H3K27Ac and suppressed H3K9me3 at the promoters of targeted inflammatory and antimicrobial genes. This leads to a shift in cellular metabolism, which is a key process in the induction of different inflammatory phenotypes in innate immunity. However, unlike the long-term effects observed in adults, epigenetic changes in newborns appear transient, suggesting incomplete modifications. It is unclear whether chromatin inaccessibility is a cause or a consequence of metabolic/transcriptional constraints. Metabolic reprogramming may also be limited in neonates due to developmental differences in mitochondrial function, reduced glycolytic capacity or lower metabolite availability. 58 It is likely that no single mechanism fully explains the lack of trained immunity in neonates. Instead, PRR signalling thresholds, chromatin accessibility and metabolic constraints appear to act together to limit the epigenetic and functional reprogramming required for trained immunity.
Our findings align with neonatal tolerogenic processes, particularly during early postnatal microbial colonization, which prevent excessive inflammation.59,60 These observations are also consistent with the Developmental Origins of Health and Disease (DOHaD) concept, which states that early-life exposures induce long lasting epigenetic changes that affect immune function and disease susceptibility.61,62 Adverse gestational environments, including maternal immune activation, metabolic dysregulation, or poor nutrition can persistently alter offspring immune responses and increase susceptibility to inflammatory and immune-mediated diseases.62,63 Such early-life exposures may not only compromise effective immune defense but also predispose offspring to dysregulated inflammation and disease across the lifespan. 64
In conclusion, while boosting the innate immune system of neonatal mice to strengthen the immune response in adulthood seemed promising, our findings demonstrate that adult TI protocols using MDP, CL429 and β-glucan were not adapted to neonatal mice. They also highlight the potential risks of early life immunostimulation. More research is needed to understand the effects and mechanisms of immunostimulants administered during the neonatal period and bring a general conclusion about the feasibility of using synthetic PRR agonists to safely induce TI in neonates.
In future, neonatal-specific TI strategies that consider PRR agonists specificity, developmental immunology, microbiota interactions, and epigenetic plasticity will be essential to safely harness innate memory in early life.
Supplemental material
Supplemental material - Trained immunity agonists given to neonatal mice fail to protect against leptospiral infection in adulthood and may cause adverse effects
Supplemental material for Trained immunity agonists given to neonatal mice fail to protect against leptospiral infection in adulthood and may cause adverse effects by Mégane Fernandez, Frédérique Vernel-Pauillac, Tiffany Pezier, Sonia Lacroix-Lamandé & Catherine Werts in Innate Immunity.
Supplemental material
Supplemental material - Trained immunity agonists given to neonatal mice fail to protect against leptospiral infection in adulthood and may cause adverse effects
Supplemental material for Trained immunity agonists given to neonatal mice fail to protect against leptospiral infection in adulthood and may cause adverse effects by Mégane Fernandez, Frédérique Vernel-Pauillac, Tiffany Pezier, Sonia Lacroix-Lamandé & Catherine Werts in Innate Immunity.
Footnotes
Acknowledgments
We express our sincere gratitude to Nathalie Lallier for her dedicated assistance with the mouse experimentation carried out at the INRAE Centre–Val de Loire. We also thank Fanny Faurie, Thierry Chaumeil, Emilie Lortscher, and Corinne Beaugé at INRAE-PFIE for their essential contributions to the rearing and provision of mice. Our appreciation extends to Ophélie Godefroy and Aurélia Dujardin from the Institut Pasteur Center for Animal Resources and Research (C2RA) for their support throughout this study, including animal supply and neonatal treatments. We further acknowledge the UTechS Photonic BioImaging (Imagopole), C2RT, Institut Pasteur, supported by the French Government’s Investissement d’Avenir programs Laboratoire d’Excellence Integrative Biology of Emerging Infectious Diseases (grant ANR-10-LABX-62-IBEID) and the Infrastructure d’Avenir en Biologie Santé France Life Imaging (grant ANR-11-INBS-0006), for granting us access to the IVIS Spectrum imaging system (Xenogen Corp., Alameda, CA). All experimental timelines and figure 8 were created with BioRender.com. English-language editing was performed using the DeepL Write software. The graphical abstract was created using Biorender.
ORCID iDs
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the French Institut Carnot France Futur Elevage (INRAE) (ANR 20 CARN 0012_01) and Microbes Santé (Institut Pasteur) (ANR 20 CARN 0023-01) to Sonia Lacroix-Lamandé and Catherine Werts, respectively. Mégane Fernandez was the grateful recipient of a Ph.D. grant from région Centre-Val de Loire at INRAE followed by a postdoctoral salary funded by an International Coordination of Research on Infectious Animal Diseases (ICRAD) (grant ANR 22 ICRD 0004 01) awarded to Catherine Werts at Institut Pasteur.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.