
Abstract
Bronchial cell pyroptosis and IL-17 respectively contribute- to the pathogenesis of steroid-insensitive asthma. In this study, we aim to explore the relationship between bronchial cell pyroptosis and Th17 in airway inflammation of steroid-insensitive asthma. The steroid-insensitive asthma model of mice was induced by toluene diisocyanate (TDI), which was also intraperitoneally injected with NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor MCC950. The bronchial epithelial cell pyroptosis was identified in morphology by transmission electron microscope. Protein expressions of pyroptosis cytokines (pro-Caspase-1, Caspase-1 p20, pro-GSDMD, cleaved-GSDMD and HMGB1), IL-17A, IL-17F and phosphorylated STAT3 (p-STAT3) in lung tissues were assessed by western blotting. Th17 in lung tissues was measured by flow cytometry. IL-17A + and p-STAT3 + cells in airway were identified by immunohistochemistry. In steroid-insensitive asthma mice, bronchial epithelial cell pyroptosis was confirmed in morphology using transmission electron microscope. Compared with controls, the protein expressions of Caspase-1 p20, cleaved-GSDMD and HMGB1 in lung tissues were increased in mice with steroid-insensitive asthma, which could be attenuated by MCC950. Th17 cells precentage and proteins expressions of p-STAT3, IL-17A and IL-17F were also increased in lung of steroid-insensitive asthmatic mice, which were also attenuated by MCC950. Similarly, the counts of IL-17A + cell and p-STAT3 + cell were more in airway of steroid-insensitive asthmatic mice than controls, and was attenuated by MCC950. In conclusion, bronchial epithelial cell pyroptosis could promote Th17 inflammation in airway of steroid-insensitive asthma mouse, which will provide further understanding on the interaction between innate immunity and acquired immunity in the pathogenesis of steroid-insensitive asthma.
Introduction
Glucocorticosteroids, also called glucocorticoids, corticosteroids or steroids, are the first-line therapy in asthma. However, 5–10% of the asthmatic population respond poorly to a high-dose of inhaled glucocorticoids, and then systemic steroids. 1 Those patients with the steroid-insensitive asthma form a category of severe asthma. It is associated with poor quality of life and an increase of morbidity and mortality, and constitutes a major societal and health care burden. 1 Possible mechanisms for steroid-insensitive asthma may be related to immune dysregulation in airway. 1 However, the mechanisms are still needed to clarify. In our study, we focus on the abnormal interaction between innate immunity and acquired immunity in the pathogenesis of steroid-insensitive asthma, which may provide noval direction on exploring the mechanisms of steroid-insensitive asthma.
Pyroptosis is an inflammatory mode of cell death. Upon the activation of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3), pro-Caspase-1 is activated into Caspase-1 p20. As a result, pro-IL-1β cleaves into bioactive IL-1β and gasdermin D (GSDMD) cleaves into active N-terminal fragment (GSDMD-N, or cleaved GSDMD). 2 As the pyroptotic executioner, GSDMD-N oligomerizes on the cell membrane to form pores,3,4 which leads to rapid cell rupture and then releasing IL-1β and HMGB1.5,6 Bronchial epithelial pyroptosis has been observed in murine model of steroid-insensitive asthma. 7 However, it remains underexplored in explicit relationship between bronchial epithelial pyroptosis and immune dysregulation in airway of steroid-insensitive asthma. Thus, in this study, as component of innate immunity, bronchial epithelial cell is investigated for its pyroptosis.
Interestingly, HMGB1 could promote Th17 cell differentiation and IL-17 production in asthma.8,9 Patients with steroid-insensitive asthma are showed more Th17 cells and higher IL-17A levels than controls. 10 In mice model, Th17 response is increased in steroid-insensitive asthma. 11 The adoptive transfer of Th17 cells could lead to steroid insensitivity. 12 Thus, Th17 cells could promote airway hyperresponsiveness (AHR) and steroid-insensitive inflammation in asthmatic mice. 12 Th17 cells contribute to the development of steroid-insensitive asthma via producing IL-17A and IL-17F. 13
In the immune pathogenesis of steroid-insensitive asthma, Th17 may be a hub through which innate and acquired immune responses are interlinked. On one hand, Th17 recruits neutrophils in airway inflammation. On the other hand, Th17 cell differentiation and Th17-dominant neutrophilic airway inflammation can be modulated by lipid metabolism and mesenchymal stem cells.14-16 However, it's not clarified the interaction between Th17 response and bronchial epithelial cell. In this study, we explore the relationship between bronchial pyroptosis and Th17 inflammation in airway with murine model of toluene diisocyanate (TDI)-induced asthma. TDI-induced model of asthma in mice shows steroid-insensitive characteristics as asthmatic patients.7,17,18 This study may provide further understanding on the immune pathogenesis of steroid-insensitive asthma from the interaction between innate and acquired immune responses.
Results
The steroid-insensitive asthma mice model was established by TDI
AHR in mice was assessed using the percentage of baseline lung resistance (RL). The baseline RL was the value at 0 mg/mL methacholine. The RL did not significantly change after prednisone or fluticasone propionate treatment (Figure 1). That's the characteristic of steroid-insensitive asthma model.

Lung resistance (RL) of mice. Multiple groups were compared using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. N = 6 mice per group. TDI: toluene diisocyanate. NS: normal saline. Pre: systemic prednisone. FP: inhaled fluticasone propionate. MCC950: NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor. *: P < 0.05.
In lung tissues, the airway inflammation and thickness of the peri bronchial smooth muscle layer were much more serious in TDI-induced mice, when compared with controls. Those changes were also observed in asthmatic mice treated with fluticasone propionate (FP), systemic prednisone (Pre) or MCC950. Interestingly, the asthmatic mice with MCC950 exposure showed a trend of decrease in airway inflammation, when compared with those treated with prednisone or fluticasone propionate (Figure 2).

Pathological characteristics in the lung of mice. The airway inflammation, epithelial hyperplasia, and smooth muscle thickening were obvious in the lung tissues from TDI group, which were also shown in the asthmatic mice treated with prednisone or fluticasone propionate. Multiple groups were compared using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. (A) Control group; (B) TDI group; (C) TDI + NS group; (D) TDI + Pre group; (E) TDI + FP group; (F) TDI + MCC950 group. (G) Semiquantitative analysis of airway inflammation. (H) The thickness of the peri bronchial smooth muscle layer. TDI: toluene diisocyanate. NS: normal saline. Pre: systemic prednisone. FP: inhaled fluticasone propionate. MCC950: NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor. *: P < 0.05.
Those results suggested that TDI-induced AHR and pathological changes were not effectively treated by inhaled fluticasone propionate or systemic prednisone. Thus, steroid-insensitive asthma mice model was established by TDI.
Bronchial pyroptosis was increased in TDI-induced mice and was inhibited by NLRP3 inhibitor
The morphology of bronchial pyroptosis was observed with transmission electron microscope (Figure 3). The pyroptosis bodies in bronchial epithelial cells were significant in TDI-induced mice. In TDI group, bronchial epithelial cells were swelling, cell membranes were discontinuous, pyroptosis bodies overflowed from cells, and the mitochondria damaged. Those morphological characteristics in TDI group were similar with TDI + normal saline (NS) group, TDI + Pre group and TDI + FP group. However, the morphology of bronchial epithelial cells pyroptosis was not so serious in TDI + MCC950 group as other groups sensitized with TDI.

Bronchial pyroptosis with transmission electron microscope in the lung of mice. (A) Control group; (B) TDI group; (C) TDI + NS group; (D) TDI + Pre group; (E) TDI + FP group; (F) TDI + MCC950 group. Red arrow: the pyroptosis-related vacuolization structures. Yellow arrow: the mitochondria. Blue arrow: the injury in nuclear membrane of nucleus. TDI: toluene diisocyanate. NS: normal saline. Pre: systemic prednisone. FP: inhaled fluticasone propionate. MCC950: NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor.
As Figure 4A and B illustrated, when compared with control group, the protein expressions of activated Caspase-1 (Caspase-1 p20), cleaved GSDMD and HMGB1 in lung tissues were increased in TDI group, TDI + NS group, TDI + Pre group and TDI + FP group. In contrast, the protein expressions of activated Caspase-1 (Caspase-1 p20), cleaved GSDMD and HMGB1 in lung tissues from TDI + MCC950 group were lower than that in TDI group or TDI + NS group.

Protein expression of pyroptosis-related cytokines and Th17 factors in the lung of mice. Multiple groups were compared using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. N = 6 mice per group. (A) Western blotting; (B) Quantitation of protein bands showing protein expression of pro-Caspase-1, Caspase-1-p20, pro-GSDMD, cleaved GSDMD and HMGB1; (C) Quantitation of protein bands showing protein expression of phosphorylated STAT3, IL-17A and IL-17F. p-STAT3: phosphorylated STAT3. TDI: toluene diisocyanate. NS: normal saline. Pre: systemic prednisone. FP: inhaled fluticasone propionate. MCC950: NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor. *: P < 0.05.
In summary, those results showed significant pyroptosis in TDI group, TDI + NS group, TDI + Pre group and TDI + FP group, which could be attenuated by MCC950.
Th17 was activated in TDI-induced mice and was attenuated by NLRP3 inhibitor
The flow cytometry showed that the percentage of Th17 cell in lung CD4 + cells were significantly increased in TDI group (TDI group vs Controls: 1.92%±0.18% vs 0.98%±0.21%, P < 0.05), which was similar with that in TDI + Pre group (1.78%±0.27%), or TDI + FP group (1.81%±0.27%). In contrast, Th17 cell percentage was decreased in TDI + MCC950 group when compared with TDI group (1.39%±0.19% vs 1.92%±0.18%, P < 0.05) (Figure 5).
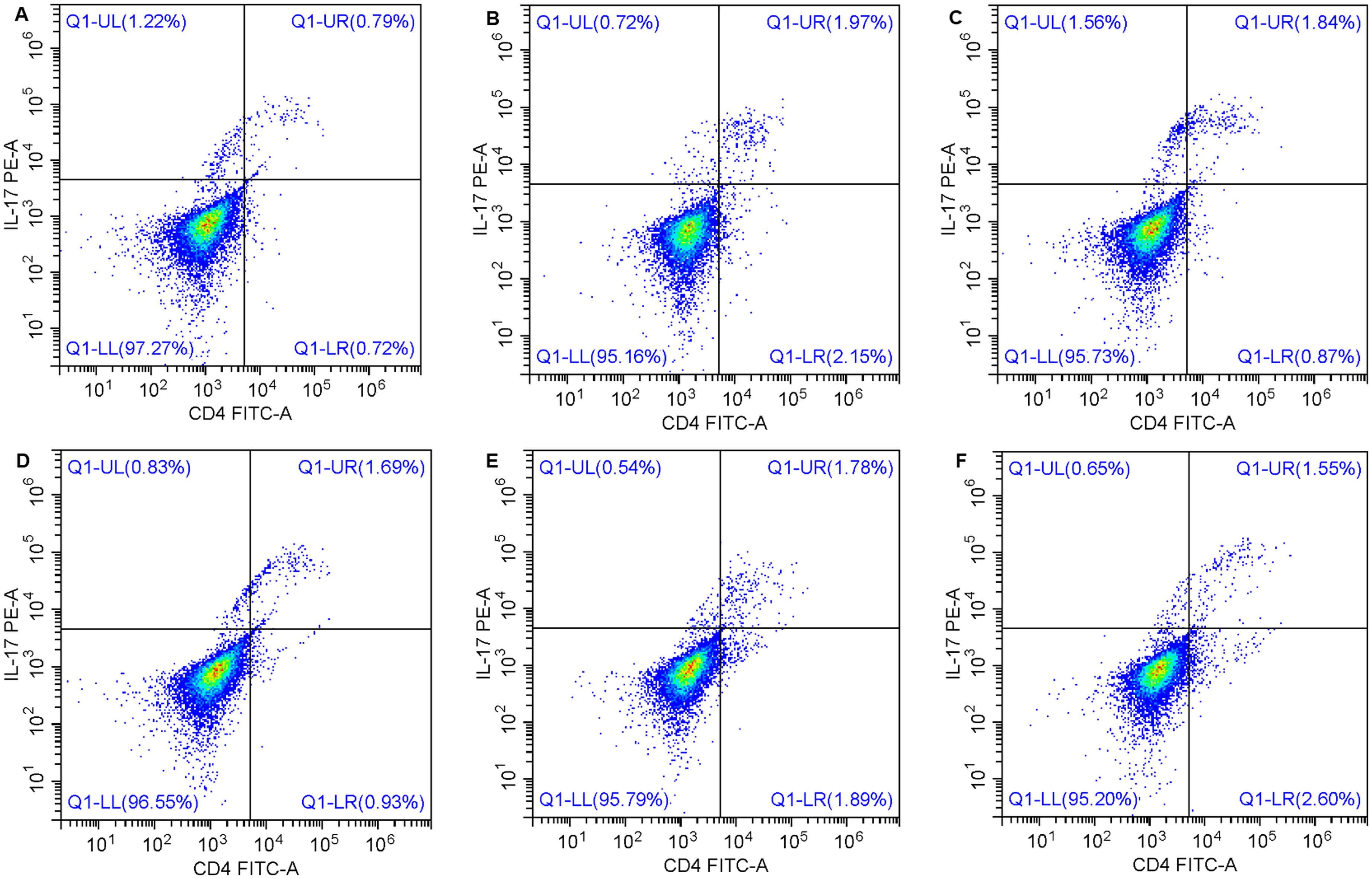
Th17 percentage in the lung of mice by flow cytometry. Multiple groups were compared using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. N = 6 mice per group. (A) Control group; (B) TDI group; (C) TDI + NS group; (D) TDI + Pre group; (E) TDI + FP group; (F) TDI + MCC950 group. TDI: toluene diisocyanate. NS: normal saline. Pre: systemic prednisone. FP: inhaled fluticasone propionate. MCC950: NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor.
When compared with controls, the protein expressions of phosphorylated STAT3 (p-STAT3), IL-17A and IL-17F were significantly increased in lung tissues from TDI group, TDI + NS group, TDI + Pre group and TDI + FP group, which could be attenuated by MCC950 (Figure 4A,C). In similar, p-STAT3 + cell and IL-17A + cell were also more in lung tissues from TDI group, TDI + NS group, TDI + Pre group and TDI + FP group than controls, which was also attenuated by MCC950 (Figure 6).

The expression of Th17-related cytokines in the lung of mice. Multiple groups were compared using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. (A) phosphorylated STAT3 + cell; (B) IL-17A + cell. TDI: toluene diisocyanate. (C) Negative control for STAT3 + cell. (D) Negative control for IL-17A + cell. (E) Quantification of phosphorylated STAT3 + cell. (F) Quantification of IL-17A + cell. NS: normal saline. Pre: systemic prednisone. FP: inhaled fluticasone propionate. MCC950: NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inhibitor. Red arrow: positive cells. *: P < 0.05.
Those results showed that Th17 differentiation and activation were increased in TDI group, TDI + NS group, TDI + Pre group and TDI + FP group, which could be attenuated by MCC950.
Discussion
This study shows bronchial epithelial cell pyroptosis in steroid-insensitive asthma of mice, and an increase of Th17 cell differentiation and activation in airways. These results suggest that bronchial pyroptosis may promote Th17 inflammation in airway of steroid-insensitive asthma.
In this study, the murine steroid-insensitive asthma model was established by TDI according to previous reports,7,17,18 since TDI-induced model showed Th17 inflammation in lung and bronchial epithelial pyroptosis.7,14 The AHR and pathological injury in airway were not suppressed by inhaled fluticasone propionate or systemic prednisone in our study, demonstrating the establishment of steroid-insensitive asthma model in mice.
In our study, the bronchial epithelial cell pyroptosis in steroid-insensitive asthma model was confirmed from the morphological characteristics using transmission electron microscope. Moreover, the protein expressions of pyroptosis cytokines, namely Caspase-1-p20 and cleaved GSDMD, were detected in lung tissues. Our results showed those expressions of cytokines were increased in steroid-insensitive asthmatic mice, which could be attenuated by NLRP3 inhibitor. Those results were consistent with findings in previous report, 7 and further confirmed the bronchial epithelial cell pyroptosis in steroid-insensitive asthmatic mice. The NLRP3 mediates neutrophilic airway inflammation in severe asthma. 19 NLRP3 is crucial in pathways which are related to pyroptosis in severe asthma of both asthmatic patients and murine model. It could promote the asthma when it's up-regulated. 20 The activation of NLRP3-Caspase 1 p20-cleaved GSDMD pathway in airway epithelium pyroptosis promotes asthma. 21 Our study observed similar findings in steroid-insensitive asthma model, and we further investigated the effect of the NLRP3- caspase 1 p20-cleaved GSDMD pathway in airway epithelium pyroptosis.
Moreover, the N-terminal pore-forming GSDMD fragment (GSDMD-N or cleaved GSDMD) could damage both inner and outer mitochondrial membranes and promote pyroptosis. 22 Thus, the cleaved GSDMD could be a marker of pyroptosis. The increased expression of cleaved GSDMD in our study demonstrated the bronchial pyroptosis in lung tissues of steroid-insensitive asthma model. Interestingly, the GSDMD-N could lead to a release of HMGB1 in pyroptosis. 6 Previous study found that in TDI-induced asthmatic mice, TDI could induce nucleocytoplasm translocation of HMGB1 and promote its release in the broncho-alveolar lavage fluid (BALF). 23 Our study not only confirm that findings, but also investigated the potential mechanism of HMGB1 up-regulation and its effect on bronchial immune-inflammation.
In the present study, the results showed an increase of Th17 cells percentage and expression of p-STAT3, IL-17A, IL-17F proteins in the lung of steroid-insensitive asthmatic mice, which was consistent with previous report. 18 Th17 cells could mediate granulocytic airway inflammation in steroid-insensitive asthma. 24 Furthermore, the increase of Th17 response could be attenuated by NLRP3 inflammasome inhibitor. Those results demonstrated that pyroptosis could promote Th17 response in the lung of steroid-insensitive asthma. HMGB1 could promote Th17 cell differentiation and IL-17 production in asthmatic mice, including TDI-induced steroid-insensitive asthma.8,9,18 Thus, our results suggested that bronchial epithelial cell pyroptosis may promote Th17 cell differentiation and activation via HMGB1 in steroid-insensitive asthmatic mice.
Th17 dysregulation plays more important role than Th2 response in severe asthma and steroid-insensitive asthma, since IL-17 could promote neutrophilic inflammation in airway.25,26 Thus, Th17 immune response has been therapeutic target in steroid-resistant asthma.27,28 Our study explored the potential mechanism with Th17 from the interaction between innate immunity and acquired immunity. And we found that the structural cell in airway, namely bronchial epithelium, may be an up-stream to promote Th17 differentiation. Those findings could provide further understanding on the interaction between bronchial epithelium in innate immunity and Th17 immune-inflammation in acquired immunity. Moreover, previous study reported that IL-17A could reduce histone deacetylase (HDAC) activity in bronchial epithelial cells, and appeared to be involved in IL-17A-induced steroid insensitivity. 25 Thus, Future study would explore mechanisms of the interaction between bronchial epithelial cell pyroptosis and Th17 inflammation from HDAC activity in steroid-insensitive asthma.
In our study, female mice were used to establish asthmatic model. Since asthma is a sexually dimorphic disease with greater documented prevalence and severity in women compared to men, female mice are more common than males in the development of asthmatic model. Female mice were demonstrated to establish different experimental asthma models, not only allergic asthma model, but also steroid-insensitive asthma. 29 Thus, female mice were used in our study. While asthmatic model of female mice had greater adaptive responses (T and B cells), male data suggested a stronger innate immune response. 30 Thus, our results based on female mice may be more applicable in female patients with asthma than the males.
In conclusion, bronchial epithelial cell pyroptosis could promote Th17 inflammation in airway of steroid-insensitive asthma mouse, which will provide further understanding on the interaction between innate immunity and acquired immunity in the pathogenesis of steroid-insensitive asthma. Future research may be focused on the pathway through which bronchial epithelial cell pyroptosis promotes Th17 inflammation in vitro.
Methods
Experimental animals
Female BALB/c mice at the age of 6–8 weeks old were purchased from Hunan SJA Laboratory Animal Company (Changsha, China). Animals were housed in standard conditions (12 h/12 h dark/light cycle, 20–25°C temperature, and 45–65% humidity) and free accessed to food and water.
Study design
Mice were randomized to each group on random numbers by a researcher who was blinded to judge the results. TDI-induced asthmatic model was established and challenged according to previous reports.7,17,18 Briefly, the mice were sensitized with 0.3% TDI at the back of both ears (20 μL for each ear) on day 1 and day 8. On day 15, 18 and 21, the mice nebulized inhaled 3% TDI (T39853, Sigma, MA, USA) for 3 h each time. The mice in control group were sensitized and challenged with the same amount of vehicle. For the TDI + Pre group or TDI + FP group, on day 15, after inhalation of TDI, the mice were intraperitoneally injected with prednisone (B2148, APExBIO, TX, US) at the dose of 5 mg/kg in TDI + Pre group, or were treated with intranasal fluticasone propionate (S31577, Shanghai yuanye Bio-Technology Company, Shanghai, China) at the dose of 300 μg/kg in TDI + FP group. The treatment of prednisone or fluticasone propionate was one time in each day and lasted 7 days. For the TDI + MCC950 group or TDI + NS group, on the basis of TDI challenge, since day 14, the mice were intraperitoneally injected with NLRP3 inhibitor MCC950 (B7946, APExBIO, TX, US) or normal saline at the dose of 10 mg/kg every other day until day 21. Six mice were included in each group. Experiments were repeated three times.
Ethical statement
All animal experiments were approved by the Institutional Animal Care and Use Committee of Guilin Medical University (approval number GLMC202105082), and according to the ARRIVE guideline.
Airway responsiveness measurements
As previously described, 18 AHR was assessed by RL measurement (Buxco Electronics, NY, USA) on day 22 when 24 h after the last administration. The response to aerosolized methacholine at the dose of 6.25, 12.5, 25 and 50 mg/mL was observed. The percentage of baseline (RL at 0 mg/mL methacholine) for each concentration of methacholine was presented as AHR.
Histology and immunohistochemistry staining for lung tissues
Mice were anaesthetized using 2% isoflurane inhalation and killed by cervical dislocation. Formalin-fixed and paraffin-embedded lung tissues of mice were cut in sections, which were stained with hematoxylin and eosin (HE). The airway inflammation was quantified according to peri bronchial inflammation as previously described. 31 In brief, the severity of inflammation was graded in five categories: 0, normal; 1, few inflammatory cells; 2, a ring of inflammatory cells 1 cell layer deep; 3, a ring of inflammatory cells 2–4 cells deep; 4, a ring of inflammatory cells of >4 cells deep. The thickness of the peri bronchial smooth muscle layer in large airways was measured from the innermost aspect to the outermost in the circumferential smooth muscle layer. 32
For immunohistochemistry, lung-tissue sections were incubated with primary rabbit anti-mouse antibody against p-STAT3 (dilution 1:500, E121-31; Abcam, Cambridge, UK), or IL-17A (1:200, 26163-1-AP; Proteintech, IL, USA) for overnight at 4°C, and then were incubated for 30 min at 37°C with horseradish peroxidase conjugated anti-rabbit IgG antibody and subsequent rinsed in PBS three times for 5 min. 3′3-diaminobenzidine-tetrahydrochloride was applied as a chromogen for 1–5 min. Sections were counterstained in haematoxylin for 5–10 min. Micrographs were obtained in optical microscope (BA410 T, motic, Xiamen, China).
The lung tissues were fixed by glutaraldehyde and osmic acid, and then embedded by epoxy resin. The ultrathin sections were doubly stained with uranyl acetate and lead curate. The bronchial pyroptosis was characterized by transmission electron microscopy (JEM1400, JEOL, Japan).
Western blotting
The lung tissues were treated with 300 μl RIPA for 10 min on ice, and then were centrifuged at 12,000×g (4°C) for 15 min. Cells were lysed with 1× RIPA Lysis Buffer (EMD Millipore, Billerica, MA, USA). The loaded proteins (150–250 μg) were separated on a 10% SDS-PAGE, followed by transferring onto NC membranes. The samples were blocked with PBST containing 5% skim milk for 90 min at room temperature. The membranes were incubated with mouse anti-mouse antibody against mouse caspase-1 (dilution 1:1000, AG-20B-0042-C100; Biomol, Hamburg, Germany), rabbit anti-mouse antibody against GSDMD (1:1000, ab209845; Abcam, Cambridge, UK), HMGB1 (1:15000, 10829-1-AP; Proteintech, IL, USA), p-STAT3 (1:2000, E121-31; Abcam, Cambridge, UK), IL-17A (1:1000, 26163-1-AP; Proteintech, IL, USA), or IL-17F (1:1000, A6487; ABclonal, Wuhan, China) and mouse anti-mouse antibody against β-actin (1:5000, 60008-1-Ig; Proteintech, IL, USA) at 4°C overnight, then 30 min at room temperature. And then they were incubated with horseradish peroxidase conjugated goat anti-rabbit (1:5000, AWS0002; Abiowell, Changsha, China) or anti-mouse antibody (1:5000, AWS0001; Abiowell, Changsha, China) for 90 min at room temperature. Blots were finally developed with the ECL Plus reagents (Thermo pierce, IL, USA). The protein bands were analyzed using Quantity One software (Bio-Rad, CA, USA).
Flow cytometry analysis
The lung tissues were disrupted by collagenase I (0.2 mg/mL) to collect single cell suspensions. Cells were stimulated with cell stimulation cocktail (00-4975-93, eBioscience, CA, USA) at 37°C for 5 h, and then incubated with IC fixation buffer (420801, Biolegend) for 30 min without light according to manufacturer's protocol. The cells were stained with CD4-FITC (11-0041-82, eBioscience, CA, USA) and IL-17A-PE (12-7177-81, eBioscience, CA, USA) for 30 min from light. After washing, cells were analyzed on a flow cytometer (A00-1-1102, Beckman, CA, USA). The gating method was negative control which wasn't labeled with fluorescence.
Statistical analysis
Group data were expressed as the mean ± standard deviation (SD). Multiple groups were compared using one-way analysis of variance (ANOVA) followed by Bonferroni post hoc test. P values <0.05 were considered statistically significant. All statistical analyses were performed using SPSS 21.0 (IBM SPSS Inc., Chicago, IL, USA).
Supplemental Material
sj-jpg-1-ini-10.1177_17534259251372592 - Supplemental material for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse
Supplemental material, sj-jpg-1-ini-10.1177_17534259251372592 for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse by Yun Lin, Jianhua Yin, Xia Yang, Jianghong Wei, Yaxi Liang, Chengfeng Zhou, Dongfang Zou and Shuyuan Chu in Innate Immunity
Supplemental Material
sj-jpg-2-ini-10.1177_17534259251372592 - Supplemental material for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse
Supplemental material, sj-jpg-2-ini-10.1177_17534259251372592 for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse by Yun Lin, Jianhua Yin, Xia Yang, Jianghong Wei, Yaxi Liang, Chengfeng Zhou, Dongfang Zou and Shuyuan Chu in Innate Immunity
Supplemental Material
sj-rar-3-ini-10.1177_17534259251372592 - Supplemental material for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse
Supplemental material, sj-rar-3-ini-10.1177_17534259251372592 for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse by Yun Lin, Jianhua Yin, Xia Yang, Jianghong Wei, Yaxi Liang, Chengfeng Zhou, Dongfang Zou and Shuyuan Chu in Innate Immunity
Supplemental Material
sj-docx-4-ini-10.1177_17534259251372592 - Supplemental material for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse
Supplemental material, sj-docx-4-ini-10.1177_17534259251372592 for Bronchial pyroptosis promotes Th17 inflammation in steroid-insensitive asthma mouse by Yun Lin, Jianhua Yin, Xia Yang, Jianghong Wei, Yaxi Liang, Chengfeng Zhou, Dongfang Zou and Shuyuan Chu in Innate Immunity
Footnotes
Author contribution
YL, JY and SC conceived the study. YL, XY, JW, YL and SC contributed to the experiments. SC wrote the manuscript. All authors revised the manuscript.
Funding
This work was supported by grants from the Guangxi Natural Science Foundation (No. 2022GXNSFAA035626) in China, Guangxi Key Research and Development Plan (Guike AB24010096), Guangxi Science and Technology Base and Talent Special Project (Guike AD24999032), and the Promotion Project of Scientific Research Ability of Young and Middle-aged Teachers in Guangxi Universities in 2023 (No: 2023KY0539) in China, and Guangxi Medical and Health Key Discipline Construction Project in China.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.