
Abstract
At the end of 2019, an outbreak of a severe respiratory disease occurred in Wuhan China, and an increase in cases of unknown pneumonia was alerted. In January 2020, a new coronavirus named SARS-CoV-2 was identified as the cause. The virus spreads primarily through the respiratory tract, and lymphopenia and cytokine storms have been observed in severely ill patients. This suggests the existence of an immune dysregulation as an accompanying event during a serious illness caused by this virus. Natural killer (NK) cells are innate immune responders, critical for virus shedding and immunomodulation. Despite its importance in viral infections, the contribution of NK cells in the fight against SARS-CoV-2 has yet to be deciphered. Different studies in patients with COVID-19 suggest a significant reduction in the number and function of NK cells due to their exhaustion. In this review, we summarize the current understanding of how NK cells respond to SARS-CoV-2 infection.
SARS-CoV-2 overview
At the end of 2019, an alert was raised in Wuhan China due to an increase in cases and deaths caused by pneumonia of unknown etiology. Later, in early 2020, genome sequencing allowed the identification of the causative agent as a virus of the coronavirus family which was named SARS-CoV-2. 1 This virus is the cause of the pandemic disease called Covid-19 and it belongs to the Betacoronavirus genus its genome is 79% similar to the SARS-CoV of 2002 and 50% to the MERS-CoV of 2012. 2
The SARS-CoV-2 sequence is 96.2% identical to the RaTG13 genome in bats. 1 However, its receptor binding domain in the S protein is very similar to that found in pangolins, 3 suggesting bats as the main reservoirs and pangolins as intermediate hosts.
Coronaviruses are positive sense single-stranded RNA (ssRNA) viruses, which under the electron microscope are observed as spherical entities with an envelope with projections of glycoproteins, giving it the appearance of a crown. 4 Like the other coronaviruses, SARS-CoV-2 structurally consists of 4 proteins, the spike (S), the envelope (E), the membrane (M), and the nucleocapsid (N) protein, being the S protein the one associated with the pathogenesis of the virus due to its binding domain to the receptor expressed in the host cells.5,6 Due to the high affinity of S glycoprotein to the receptor for angiotensin convertase enzyme 2 (ACE2), which is found in the epithelium of the upper and lower respiratory tract and in lung tissue,7,8 SARS-CoV-2 causes respiratory tract infections that can result in pneumonia with serious complications and mortality.9,10
Epidemiology and clinical picture (COVID-19)
The animal-human transmission and the adaptation of the virus was what gave rise to this outbreak; 11 however, its transmission from human-human occurs by air when an infected person speaks, sneezes, or coughs, and this has become a global health crisis.12,13 Since the first cases reported in Wuhan, the virus has spread in more than 200 countries, confirming to date 236.77 million cases and 4.83 million deaths worldwide, with a global fatality rate of 2.04%. 14
To date, 3,720,545 cases and 281,958 total deaths have been confirmed by COVID-19 in Mexico, with an incidence of 2884.8 accumulated cases per 100,000 inhabitants. 15 The distribution of cases by sex shows a predominance of 50.1% in women, with a median age of 39 years. Only 10 entities from the 32 states accumulate the highest number of cases in the country, making up for more than two thirds (66%) of all the accumulated cases in the country. 15
Regarding deaths in México, the distribution by sex in confirmed deaths shows a prevalence of 62% in men, with a median age of 64 years. The accumulated mortality rate per week in México to date is 218.6 per 100,000 inhabitants. 15
The clinical spectrum of SARS-CoV-2 infection ranges from asymptomatic infection to critical illness and death. Among patients who are symptomatic, the symptoms appear between 4 and 5 days after contact with the virus and they will present all the symptoms after 12 days.16,17 The time between the onset of the symptoms and the death of patients with critical illness is from 6 to 41 days, with a mean of 14 days. 18 However, the time of appearance of the symptoms, as well as the severity of the disease, will depend on the age of each patient and the state of the patient's immune system.18,19 Some risk factors that worsen the disease are immunodeficiency, cardiovascular disease, chronic lung disease, diabetes, and obesity.20,21
The most common symptoms are fever, cough, myalgia, and fatigue. However, patients can also present sputum production, headache, and gastrointestinal problems, such as diarrhea and nausea.22,23 The severity of the disease leading to fatality is associated with the progression of symptoms towards respiratory distress, a decrease in oxygen saturation, and the development of acute respiratory distress syndrome (ARDS). 22
Lymphopenia and elevated levels of lactate dehydrogenase, C-reactive protein, ferritin, prolonged prothrombin time, elevated levels of liver enzymes, as well as D-dimer are among the laboratory findings of hospitalized patients.22,24,25
Pathogenesis and immune response
Infected patients can develop acute respiratory distress and respiratory failure as the leading cause of death, but damage to other organs and systems, including heart, liver, and kidney, also contribute to death. These damages can be attributed to a hyperinflammatory innate immune response along with an inadequate adaptive response that lead to an exacerbated and uncontrolled immune response, in which a storm of inflammatory cytokines predominates, also in addition to the direct attack of SARS-CoV-2 itself, which may elicit extensive local and systemic tissue injury.26,27
Three phases have been observed in the pathogenesis of COVID-19: the viremia phase, the acute phase (pneumonia phase), and the severe or critical phase. 27 If the immune function in the acute phase is effective, the virus can be suppressed keeping inflammatory lung damage to a minimum and entering the recovery phase.28,29 If the body is unable to effectively control the virus, it will cause an overproduction of pro-inflammatory cytokines that will damage the lung infrastructure and the patient will become severely or critically ill.28,29
Since the SARS-CoV-2 virus has a receptor-binding domain similar to that of SARS in its S protein. it has been determined that the entry into the cell is through the ACE2 receptor.11,30 SARS-CoV-2 enters the respiratory tract and binds to cells presenting the ACE2 receptor and transmembrane protease serine 2 (TMPRSS2), as it occurs in the epithelial lining of the lungs.7,31
The active replication of the virus and its release generates an increase in the plasma levels of angiotensin II, which suggests an imbalance in the renin angiotensin system. 32 In turn, the cytopathic activity of the virus induces the lysis of cells, 33 which, together with angiotensin II, induces a strong immune response; the triggering of a storm of inflammatory cytokines such as IL-1β, IL-6, IP-10, TNF-α, IL-8, IFN-γ, MIP1α, MIP1β, and MCP1 has been observed in patients with moderate and severe COVID-19- These cytokines further activate immune cells and increase the pulmonary infiltration that can develop ARDS with significant lung damage that can culminate in the death of the patient.24,34
On the other hand, the early production of some cytokines, such as IL-17A and IFN type I and III, may tend to a favourable resolution of the disease, since these are increased in those patients who manage to recover without squealed, compared to the critically ill.19,35 Recently, it's been observed that the production of IFN type I and III is positively related to a low viral production 35
In the alveoli of COVID-19 patients, the infiltration of macrophage is increased. Since this cells produce cytokines such as IL-6, TNFα, and IL-10, these have been associated with severe COVID-19, for more than half of ARDS patients have manifested macrophage activation syndrome with high levels of TNF and IL-6 in the circulation. 36
The analysis of circulating immune cell populations showed that there is a prevalence of myeloid cell populations in patients with severe Covid-19, as well as an increase in the presence of immature lineages.25,37 Amongst these populations, the inflammatory monocytes are increased especially in severe covid-19 patients. In addition, the circulating neutrophils are also increased. The neutrophil/lymphocyte ratio can be a clinical inflammation biomarker; therefore, it may be an indicator of the severity of COVID-19 in the early stage of the disease.37,38
Similarly, it has been reported that total T lymphocytes, CD4 + T lymphocytes, and CD8 + T lymphocytes are reduced below the lower limit in the vast majority of patients with COVID-19, being more in severe cases than in moderate ones.39,40 It is observed that the B cell population is higher in severe cases than in moderate cases, which is considered as a compensation for the significant decrease in T lymphocytes. 41 In addition, the natural killer (NK) cell count is also below the normal limit.24,39
Regarding the functional characteristics that were found, it has been observed that, even though patients with severe COVID-19 present a reduction in the total number of T lymphocytes, the levels of Th1-type and Th2-type cytokines remain high, as well as with high cytotoxic activity,42,43 which indicates an over-activation of the T lymphocytes.
In patients with severe Covid-19, it's been observed that Th1/Th2 cytokines balance impact the disease outcome. Once the viral infection is settled, if the predominant immune response is Th1 type, the infection can be cleared and the prognosis is good. However, if the immune response leads to an over reactive Th2 response, this can lead to the cytokine storm and the prognosis gets worse.44,45 However, this remains poorly understood.
In the same way, in vitro studies with convalescent serum from COVID-19 patients and in vivo studies with non-human primates have suggested that neutralizing antibodies produced by B cells can exacerbate COVID-19 severity through antibody-dependent enhancement (ADE), and further exacerbating organ damage.46,47 However there is not still strong evidence that sustains this ADE hypothesis
These findings suggest that there is a dysregulated function of the immune system during the course of SARS-CoV-2 infection, being notable the decrease in cytokine regulation, and highlighting the importance of lymphocyte subpopulations as predictors of the severity of the disease.
Additionally, TLR pathways could also be involved in the pathogenesis of SARS-CoV-2 in the initial failure of viral clearance and in the development of the deadly clinical manifestations of severe COVID-19, essentially ARDS with fatal respiratory failure.
Toll-Like receptors and SARS-CoV-2
Toll-like receptors (TLR) are pattern recognition receptors (PRR) that belong to the innate immune system and recognize multiple pathogen-associated molecular patterns (PAMPs) from bacteria, viruses, and other pathogens.48,49 In addition, they recognize certain damage-associated molecular patterns (DAMPs) released from dying or lytic cells during host tissue injury or viral infection.48,49
The human TLR family comprises 10 members from which TLR3, 7, and 8 are essential in the recognition of RNA viruses. TLR3 sense doble-stranded RNA (dsRNA) and TLR7 and 8 senses ssRNA; recognition occurs in the endosomes of cells. 50
After TLR activation, a nuclear translocation of the NF-κB, IRF-3, and IRF-7 transcription factors develops, leading to a production of pro-inflammatory cytokines like IL-1β, IL-6, TNF-α and type I IFN, which are essential for anti-viral responses. Since it's been observed that there are high levels of IL-6 and TNF-α in patients with severe COVID-19, it is suggested that TLRs have an important role in this pathology. 51
In silico studies of molecular docking have demonstrated there is significant that the interaction between human TLRs and SARS-CoV-2 spike and E proteins suggesting that those bindings may be a step for the activation of the innate immune cells and a vital mediator for COVID-19 immunopathogenesis.52,53 In addition, there is evidence that suggests that major immunopathological consequences have resulted from the interaction between SARS-CoV-2 antigens and human TLRs.54,55 Amongst the biding between TLR and SARS-Cov-2 antigens, the bind to TLR4 was observed to be the strongest.56,57
Since TLR4 is the most efficient receptor that induces proinflammatory responses, 58 the interaction between TLR4 and the spike protein could be one of the reasons behind the immunopathological manifestation of COVID-19.
A proposed mechanism is that the virus binds TLR4 on the alveolar and bronchial epithelial cells and causes an aberrant TLR4 proinflammatory signalling. The infiltration of inflammatory cells leads to the activation of TLR4 on inflammatory cells, further exacerbating the situation as it leads to NETosis and the activation of inflammasome. The DAMPS released from necrotic/ lytic cells also activate TLR4, amplifying the already severe inflammation.57,59
A dataset analysis demonstrated that the expression of TLR2 was increased with the severity of COVID-19. In this study, it was also demonstrated that TLR2 can sense SARS-CoV-2 infection via recognition of the E protein and it is able to induce the release of inflammatory cytokines such as IL-6 and TNF-α. 60 The results of this study identified some inflammatory signalling pathways activated during SARS-CoV-2 infection, which improves the understanding of the molecular mechanism of the cytokine production.
It has also been suggested that TLR2/6 has an important role; Proud et al. revealed that the administration of TLR2/6 agonist reduces SARS–CoV–2 transmission and can protect against the development of COVID-19. When stimulated with TLR2/6 agonist, TLR2 can activate the innate immune responses such as the suppression of inflammation and avoid tissue damage as well as promotion of the integrity of the local epithelial barrier function. 61
In accordance with the docking studies, TLR3 forms a stable complex with NSP10 mRNA, and since TLR3 is most likely involved in inducing proinflammatory cytokines, the binding from SARS-CoV-2 may induce an inflammatory response through this receptor. 52
Additionally, genetic studies have provided information regarding the involvement of TLRs in the SARS-CoV-2 infection. For instance, TLR3 and TLR7 are suggested to be important in regulating the infection of cells, for it's been observed that the cells from patients with mutations on these TLR alleles are more easily infected by the virus than those without the mutation. 62 This may be due to the lack of IFN-β produced by those mutated TLR stimulation.
Another study demonstrated that TLR3 and TLR7 have different activation times, which may have an implication in the development of the disease. On one hand, TLR3 may interfere with the infection since it might act producing IFN-β 24 h post infection. On the other, TLR7 activates the NF-κB transduction pathway leading to pro-inflammatory cytokine secretion, which might lead to a cytokine storm with an adverse effect on the development of the disease. 63
A whole genome sequencing of SARS-CoV-2 revealed that it has more ssRNA motifs than SARS-CoV, and since TLR7 binds to ssRNA, it could be more involved in the pathogenesis of SARS-CoV-2.64,65 Following this speculation, the in silico data suggested a good binding for the mRNA encoding viral E-protein with the TLR7; this could activate the downstream proinflammatory signalling cascade. 52 Since the activation of TLR7 can also lead to a NET (neutrophil extracellular traps) response, binding to TLR7/8 may induce a strong proinflammatory response in patients, leading to acute lung injury; 66 therefore, it might have a dual role in the progression of the disease.
The in silico analysis also revealed that the binding of the mRNA to spike protein and E-protein to TLRs, induces a change in the conformation of the extracellular domain. 52 This change could lead to an enhanced proinflammatory function and favour the pathogenesis of COVID-19.
In the respiratory tract, several cell types express those TLR receptors and are very important for the immune response against viral infection. NKs, mucosal-associated invariant T cells, and neutrophils, alongside those TLRs play an important role during the clearance of respiratory viruses as SARS-CoV-2. 67 Therefore, a better understanding on how these receptors function their status in the different cell populations and their interaction with SARS-CoV-2 is vital for a better pathogenesis understanding and treatment approaches.
Overview of NK cells
NK cells are the effector lymphocytes of the innate immune system whose function is immunosurveillance and to exert a rapid response against infected or modified (tumour) cells.68,69 Based on the expression density of CD56 on the surface of the NKs, these can be classified into two populations, the NK CD56bright cells, which express a higher density of CD56 and have a greater capacity to produce cytokines, thus, it is considered that they are regulators of the immune response, and the NK CD56dim cells, which express CD56 in a lower density; they are poor cytokine producers and have a higher cytotoxic capacity due to having a large quantity of lytic granules.70,71
NK cells have a repertoire of germline receptors which are traditionally classified as inhibitory or activator receptors. 70 Both activators and inhibitors are from two NK receptor superfamilies: the killer cell immunoglobulin-like receptors (KIRs), such as KIR2DL, KIR3DL (inhibitors) KIR2DS, KIR3DS (activators), which recognize classical MHC-I molecules, and the lectin C-type receptors, such as CD94/NKG2A (Inhibitor) and NKG2C (activator), that primarily recognize non-classical MHC molecules, such as human leukocyte antigen (HLA)-E.72,73
Physiologically, NK cells killing activities are inhibited by interactions between the receptors and their ligands (MHC-I); the balance between activating and inhibiting signals determines whether the NK cell will attack or not. Healthy cells express enough MHC-I molecules that are recognized by inhibitory receptors as “self” and induce a strong inhibitory signal. When the cells have a malignant transformation or become infected with intracellular pathogens, they experience a reduction or loss of MHC-I expression; this phenomenon, known as missing self-recognition, transforms the cell in a potential NK-cell target. 74 Additionally, this transformed or infected cells express more molecules than can be recognized by the activator receptors. The new strong activator signals, which overpower the inhibitory signals, triggers the activation of NK cells. 75
Once activated, the NK cells can induce cell death by two primary mechanisms. One is the activation of cell death receptors in the target cell by TRAIL and FAST ligands, which activate the caspase pathway.76,77 The second mechanism, which is the predominant one, involves the release of their lytic granule's perforins and granzymes. Perforin can form a pore in the target cell promoting osmotic lysis, while granzyme B is capable of inducing apoptosis by promoting the caspase-dependent and -independent apoptosis.78,79
NK cells against virus
Compared to the normal percentage of NK cells in peripheral blood (5–20%), it has been observed that the human lung is rich in NK cells, with the CD56dim CD16 + subpopulation being the predominant one.80,81 However, despite the ability to respond to viral infections by attacking infected cells as well as the regulation of antigen presenting cells, it has been described that these are hypofunctional with respect to peripheral blood NK. This has been associated with a high concentration of resident macrophages that suppresses NK activity due to their high production of IL-6.80,82
There is evidence that NK cells are essential for the protection against viral infections, as patients with NK cell deficiencies develop viral infections that lead to death.83–85 It has also been shown that NK cells are capable of responding rapidly during the acute phase of infectious processes due to viruses such as hantavirus, tick-borne encephalitis virus, influenza A virus (IAV), and dengue virus, as well as in the case of yellow fever after immunization with the vaccine.86–88
NK cells can eliminate viral infections by the ‘missing self’ hypothesis; this means NK cells can destroy virus-infected cells that lose or down regulate the MHC class I molecules on their surfaces, allowing NK cell to target them. 74 However, there are many circumstances in which NK cells can eliminate virus-infected cells expressing MHC class I. 89 It is also known that NK cells are activated through the interplay between the inhibitory and activating receptors. 90
Since NKs do not express clonal receptors, the repertoire of these cells is formed of germline receptors. Killer immunoglobulin-like receptors (KIRs) recognize MHC class I molecules and clear acute hepatitis C virus and Epstein-Barr virus; 91 the natural cytotoxicity receptors recognize viral-derived products. In humans, NKp46 recognizes influenza virus hemagglutinin and may be involved in the resistance to these viruses; it is also critical in the response to human CMV-infected cells.92,93
NK cells can also sense stress-induced ligands through the C-type lectin-like homodimer NKG2D receptor and recognize host stress proteins induced upon viral infections. 94
In addition, NKs can be activated through TLRs; this stimulation has emerged as an important role in NK cell cytotoxicity and proinflammatory activity. In NK cells, the expression of TLR1-8 mRNA is constitutive, 95 and, in vitro, it's been demonstrated that this ligand binding of TLRs 2, 3, 5, and 9 stimulates the secretion of IFN-γ. 96
It has been demonstrated in vivo that TLR2 stimulation on NK cells increases the IFN-γ production in viral infection. 97 In the same way, TLR7 signalling plays a specific role in local and systemic NK cell activation following respiratory IAV infection, leading to IFN-γ secretion and degranulation. 88
Downstream of these activating receptors, the ligand recognition triggers an intracellular kinase cascade to transmit the activation signal, 98 releasing the cytotoxic activity of NK by direct release of lytic granules towards the target cell and also proinflammatory responses, such as an increase in the production of IFN-γ in order to clear the virus-infected cells. 98
NK cells are not only capable of directly attacking and killing infected cells through their receptors and TLRs, but the activation also influences the cellular microenvironment to enhance the adaptive responses of T cells through the secretion of cytokines. 99 Depending on the type and degree of activation of NK cells, these can function as a regulator of T cells and favour the attack and control of infectious processes or favour a pro-inflammatory environment and promote immunopathology due to infection. 99
Participation of NK cells in COVID-19
During SARS-CoV-2 infection, a reduction in immune system cells has been reported and, among them, the absolute count of NK cells in peripheral blood of patients with both moderate and severe COVID-19. It has been observed that the reduction is up to 55% of the normal number, with a range of 0.2 × 108/mm3 to 2.9 × 108/mm3, representing 2.37% of the total lymphocytes in peripheral blood.25,40,100,101
In addition, the severity of the disease is related to the depletion of NK cells, since it's been observed that NK cells are significantly reduced in patients with severe disease, compared to those with non-severe disease. 102 Regarding the NK cell subpopulations, the CD56dim/CD56bright proportions are maintained; however, the CD56dim seems to be more depleted primarily in ventilator-dependent patients, whereas the CD56bright were significantly depleted in all COVID-19 patients. 37 Similarly, a higher percentage of CD16 + populations and a lower percentage of CD16- have been observed in patients with COVID-19.101,103
Initially, the reduction in the total number of NK cells was associated with their trafficking to the lung, since two studies of single cell of immune cells carried out in bronchoalveolar lavage fluid of patients with COVID-19 suggested that the number of NK cells increased at this site of infection.104,105 However, Demaria et al. observed a decrease in mature NK cells (CD56 + CD57 + CD16 + ) in the analysis of bronchoalveolar lavage fluid of patients with ARDS, 106 which suggests that the decrease in NK cell levels observed in blood is not a consequence of their migration to the infected lungs.
The reason for the decrease in blood NKs in SARS-CoV-2 pathology is not clear yet. However, due to the important antiviral function, the probable decrease in these cells favours the progression of complications that occur in the lung.
SARS-CoV-2 infection also modulates the phenotype of NK cells, since increases of certain markers have been observed in NK cells in patients with COVID-19, such as type C lectin receptors (CD69), a marker that indicates an activation of NK cells. 107 Similarly, the upregulation of the PD-1, TIM-3, NKG2A markers has been observed, predominantly in NK CD56dim.106–108 The high densities of these receptors and the upregulation of the inhibitory checkpoint receptors such as LAG3 are compatible with a dysfunctional and exhausted phenotype due to the high hyperactivation of cells associated with the CD69 receptor and by prolonged IL-15 stimulation. In the same way, the TIM-3, PD-1, and NKG2A receptors induce an inhibition of NK cells when are in contact with their ligands.37,106,107 Since NKG2A is an inhibitor of the NK activation, when it is observed to be highly expressed in NKdim, it is suggests that a lower lytic or degranulation activity is produced against infected cells of the virus.
Similarly, Maucourant et al. reported a strong activation of NK cells, NKG2C + , in COVID-19 patients. However, the high expression of NKG2C and perforins was correlated with the severity of the disease. 103 The expression of perforins and granzyme B was found elevated mainly in CD56bright NKs and this state was directly related with IL-6 levels and multiple organ failure and inversely with the partial pressure of oxygen (PaO2) in patients with COVID-19. Indicating a pro-inflammatory state of NK cells in severe COVID-19. CD56dim NK cells expressing granzyme A were negatively related with serum IL-6 levels in COVID-19 patients. 103 The aforementioned findings on the NK subpopulations imply that the hyperinflammatory state sustained by the persistent elevation of IL-6 is probably responsible for the exhaustion of NK cells after an exaggerated activation by the virus.
Recently, it's been suggested that the KIR receptor profile expressed by the NK cell of the patients may be implicated in the severity of COVID-19. Littera et al. observed that NK cell inhibitory KIR gene receptor profile prevails in patients with SARS-CoV-2 infection and that patients with severe COVID-19 had a significantly higher frequency of KIR2DL1 and KIR2DL3 inhibitory receptors. 109 In those patients, non-functional KIR2DS4 receptors and the reduction of the gene coding for the KIR2DS2 activating receptors were also observed. 109 This predominantly inhibitory profile would promote an ongoing viral replication and more prolonged inflammatory responses.
A study in lung cells revealed that the intracellular expression of the SARS-CoV-2 S protein generates a reduction in NK degranulation; simultaneously, it induces the expression of HLA-E on the surface of lung epithelial cells. Similarly, this study revealed that there is greater modulation of the NKG2A/CD94 receptor in NK cells when the S1 protein of the SARS-CoV-2 virus (SP1) is expressed in lung epithelial cells. This showed, for the first time, that NK cells are affected by the expression of SP1 in lung epithelial cells through the HLA-E/NKG2A interaction. 110
Similarly, Liao et al. suggest in their single cell study that a highly proinflammatory macrophage microenvironment is present in the lungs of patients with severe COVID-19. 104 The high levels of IL-6 produced by these macrophages may also have an important role in NK phenotype dysregulated.
It has been observed that NK cells are functionally defective in patients with COVID-19; they have the dysfunctional phenotype of NK and NKdim cells that express a high density of NKG2A and have shown a downward response in degranulation, which is evidenced by a decreased expression of CD107a + . In the same way, they have shown to have an irregular secretion of interferon- (IFN) -γ, granzyme B, and the alpha tumour necrosis factor (TNF-α), with respect to what is observed in healthy people;111,112 this supports the inhibitory role of NKG2A in the control of NK cell activation in the presence of SARS-CoV-2. In contrast, "regulatory" NK subpopulations exhibited a greater ADCC antibody-dependent response and actively expanded in the periphery in severe COVID-19 patients.112,113 It has been shown that they are less responsive to cytokine stimulation, but they respond better to signal through the CD16 receptor, so these cells could be fighting SARS-CoV-2.
COVID-19 patients display very active NK cells accompanied by an imbalance of NK cell subsets at the beginning of the disease and in mild cases. As the disease progresses, there can be a shift toward terminal differentiation demonstrated by the expression of inhibitory receptors. This can be further associated with tissue trafficking and defective NK cell cytotoxic activity in the absence of perforin, or with the triggering of the expression of these receptors.
Regarding the production of IFN-γ from NK cells, this was reduced in patients with COVID-19, predominantly in the CD56bright cell subpopulation and it has also been observed that it is associated with a significant reduction in degranulation. 71 This means that the functions of regulatory and cytotoxic NKs are diminished. Similarly, a negative relationship was found between the degranulation frequency and IFN-γ secretion with respect to high concentrations of pro-inflammatory cytokines, such as IL-6. In patients with a severe ARDS course, high concentrations of IL-6 accompanied with low levels of expression of INF-γ and granzyme B granules were observed.101,107,111 Since interferon-γ has been shown to be a suppressor of type II inflammatory processes that lessen the effects that these cause in the lung,114,115 the reduction in IFN-γ by NK cells observed during SARS-CoV-2 infection is an important functional characteristic for the control of the viral infection.
Together, these phenotypic findings and functions on the NKs subpopulations indicate that, under conditions of exaggerated systemic inflammation such as moderate and severe COVID-19, NK cells are functionally dysregulated, which leads to their exhaustion and maintenance of the inflammatory state by reducing viral lysis, favouring the transmission and pathogenic severity during COVID-19 (Figure 1).
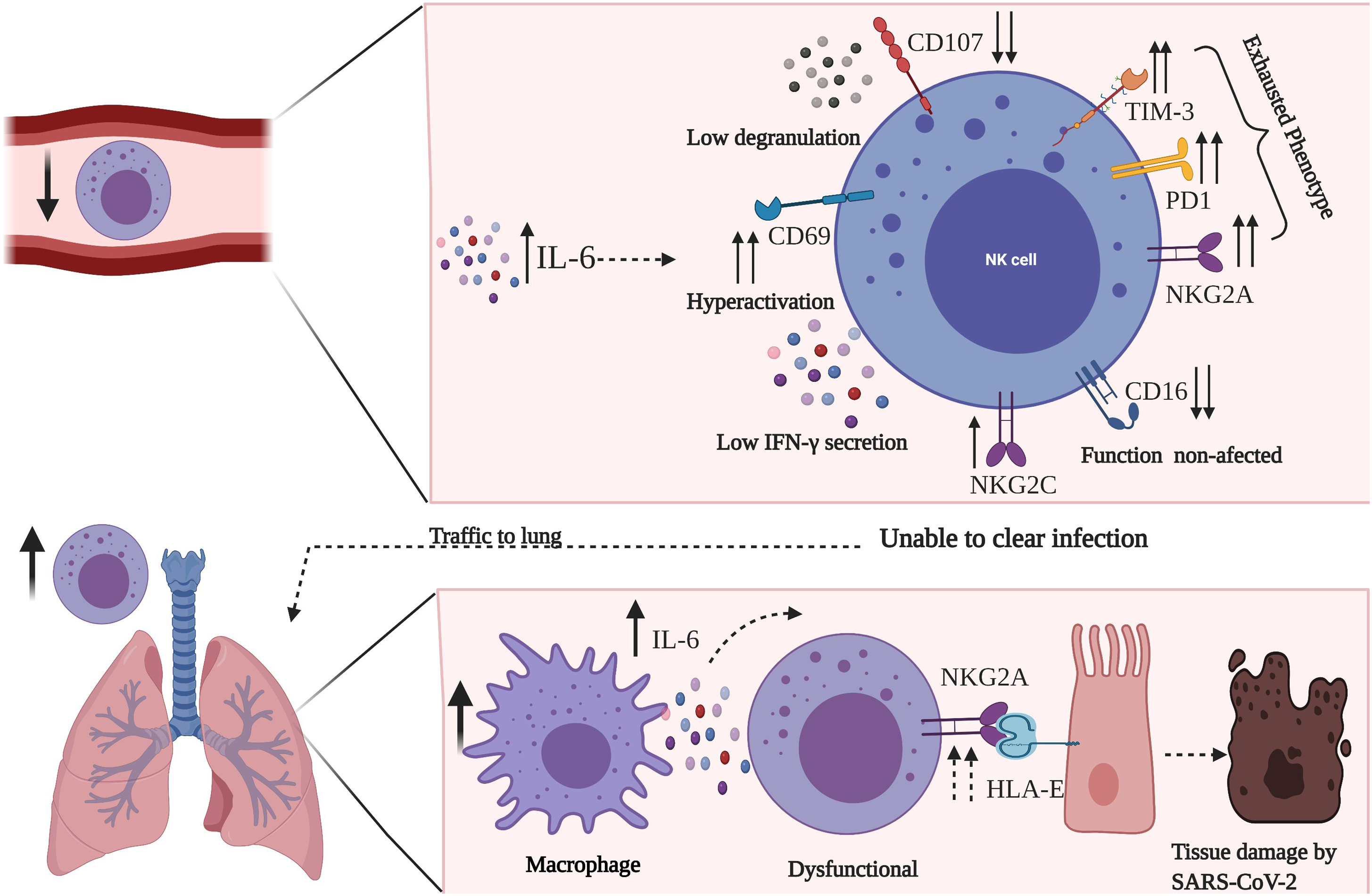
Overview of the exhausted phenotype of NK cells during the SARS-CoV-2 infection. NK cells are reduced in circulation, and express markers of exhaustion which is possibly due to high levels of IL-6. This phenotype is characterized by the high expression of TIM-3, PD-1, NKG2A, a mild expression of NKG2C and low expression of CD107. This combination resolves in a low secretion of IFN-γ and low degranulation. These cells travel to the lungs, where highly proinflammatory macrophages promote less effectively clearing of the virus-infected cells, leading an hyperinflammation and tissue damage. Importantly NK cell mechanisms are still poorly understood in COVID-19.
Until this moment, there is no information regarding the expression nor the response of TLRs in NK cells against SARS-CoV-2 infection. However, as mentioned above, these receptors are very important for a complete virus clearance by the NK, and since it is hypothesized that they have an important role in the progression of COVID-19, it is important to know the expression, regulation, and role that these receptors play in NK cells during the disease, which could help to improve the therapies.
Conclusion
NK cells are the first line of attack against viral infections; however, the results that have been obtained from the different studies with SARS-CoV-2 show exhausted NK cells with impaired functioning, mainly in patients with severe disease. This suggests that NK cells could be favouring immunopathogenesis in SARS-CoV-2 infection more than fighting it. However, the mechanisms that lead to the dysregulation of the functions of NK cells are not known yet. It is necessary to continue evaluating how to eliminate their state of exhaustion, which allows NK cells to recover their functionality and reduce the inflammatory process.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Hospital Infantil de México Federico Gómez, (grant number HIM/2019/035 SSA 1618).