
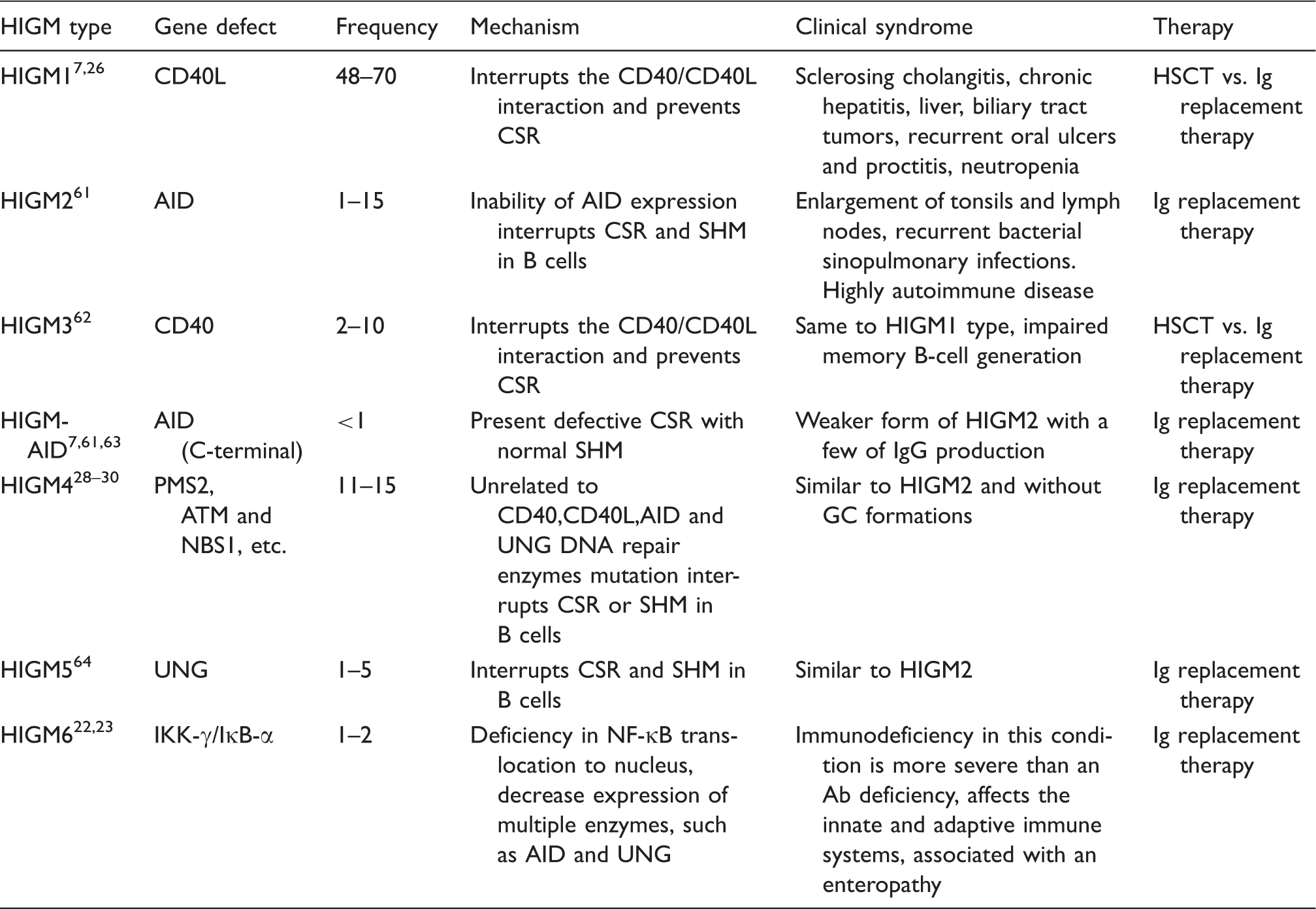
Abstract
The critical role of the CD40/CD40L pathway in B-cell proliferation, immunoglobulin (Ig) isotype switching and germinal center formation has been studied and described extensively in previous literature. Interruption of the CD40/CD40L signal causes hyper-IgM (HIGM) syndrome, which has been classified and recognized as a group of rare inherited immune deficiency disorders. Defects in CD40 and CD40L interactions or in downstream signaling molecules, including activation-induced cytidine deaminase, uracyl-DNA-glycosylase, NF-κB and DNA repair enzymes, result in an increased level of serum IgM and a significantly decreased or absent level of IgA, IgG and IgE that is accompanied by severe recurrent infections and autoimmune diseases. Many genetic defects in HIGM have been identified and, as a result, it is possible for patients to be definitively diagnosed by gene sequencing and to delineate the immunological features of the patients. Modifying the CD40/CD40L signaling pathway may offer the possibility of restoring the normal serum Ab production and curing the immunodeficiency. Hematopoietic stem cell transplantation has achieved a high rate of success using a sibling donor. In addition, successful examples of treating other immunodeficiencies using gene therapy indicated that there was a possibility of eradicating HIGM with this approach. In this review, we summarize the current drugs and a variety of therapeutic approaches for the treatment of the HIGM syndrome by interfering with the defective CD40/CD40L pathway.
Keywords
Introduction
Hyper-IgM (HIGM) is a type of rare disease that is prevalent in <1/1,000,000 of the world’s population. 1 Immunodeficiency with HIGM was first described in 1960 as a group of disorders in patients who had higher serum IgM and much lower or undetectable IgG, IgA and IgE than those of normal people. 2 This immunological phenotype is mainly due to the failure of immunoglobulin (Ig) isotype class switch recombination (CSR) and somatic hypermutation (SHM) in B cells that are associated with the CD40 and CD40 ligand (CD40L) interaction.3–7 The binding of CD40L to the CD40-induced CD40 cytoplasmic domain recruits members of the TNF receptor-associated factor (TRAF) proteins. 8 Further signaling by TRAF proteins is mediated by nuclear factor NF-κB, which activates downstream pathways leading to Ig gene switching.8,9 The interaction between CD40L on T cells and CD40 on APCs is blocked and the failure of this interaction is responsible for the immune deficiency in HIGM.
The clinical manifestations of HIGM are complex and severe. Most patients are susceptible to opportunistic and recurrent infections, and may be accompanied by autoimmune manifestations. Current clinical therapy for HIGM includes two parts: to ameliorate the clinical manifestations and to restore high-affinity Ab production. Recurrent infection is the most common manifestation of the syndrome. As a result, patients often require long-term antibiotic treatment. Meanwhile, the most efficient therapeutic approach, IgG replacement therapy, should also be applied to patients in a timely manner. However, while IgG replacement reduces the frequency and severity of infections, it does not prevent lymphoproliferative disease, sclerosing cholangitis or malignancies. 2 Gene therapy and stem-cell therapy may restore the destroyed immune system. However, owing to the complex and still unclear molecular pathology of all of the types of HIGM, stem-cell transplantation and gene therapy offer a possibility of curing only HIGM1 and HIGM3.10–12 In spite of the attempts, cured patients are rare and clinical exploration has not proceeded smoothly. The discovery of drugs that interfere with the CD40/CD40L pathway may provide HIGM patients with more convenient therapeutic options.
Molecular pathology
The classification and associative description of HIGM.
As shown in Figure 1, the binding of CD40L to the CD40-induced CD40 cytoplasmic domain recruited the members of TRAF proteins.
8
Further signaling by TRAF proteins was mediated by NF-κB, which activated downstream pathways leading to Ig gene switching.8,9 Patients with HIGM1 (also called X-linked HIGM, XHIGM) and HIGM3 have defects in the CD40L gene and CD40 receptor gene, respectively.15,16 As a result, the interactions between CD40L on T cells and CD40 on APCs are blocked, which results in the absence of CSR.
17
Brief description of the interaction of a CD4+ T cell with B cells. CD40L is transiently expressed on the surface of the CD4+ T cell after interaction between the T-cell receptor complex and MHC class II molecule. CD40L interacts with CD40 on the B cell and recruits TRAFs, leading to the expression of co-stimulatory molecules (CD80 and CD86) and IL-12 secretion, and the promotion of IFN-γ-dependent T-cell differentiation. Additionally, CD40 signaling in B cells activates NF-κB translocation to the nucleus via IKK-γ and IκB-α, leading to the expression of AID, UNG and DNA repair enzymes (latter not shown) in B cells, which results in CSR and SHM, and the generation of germinal centers and production of high-affinity Abs. The deficient molecules involved in HIGM are labeled in red and red box.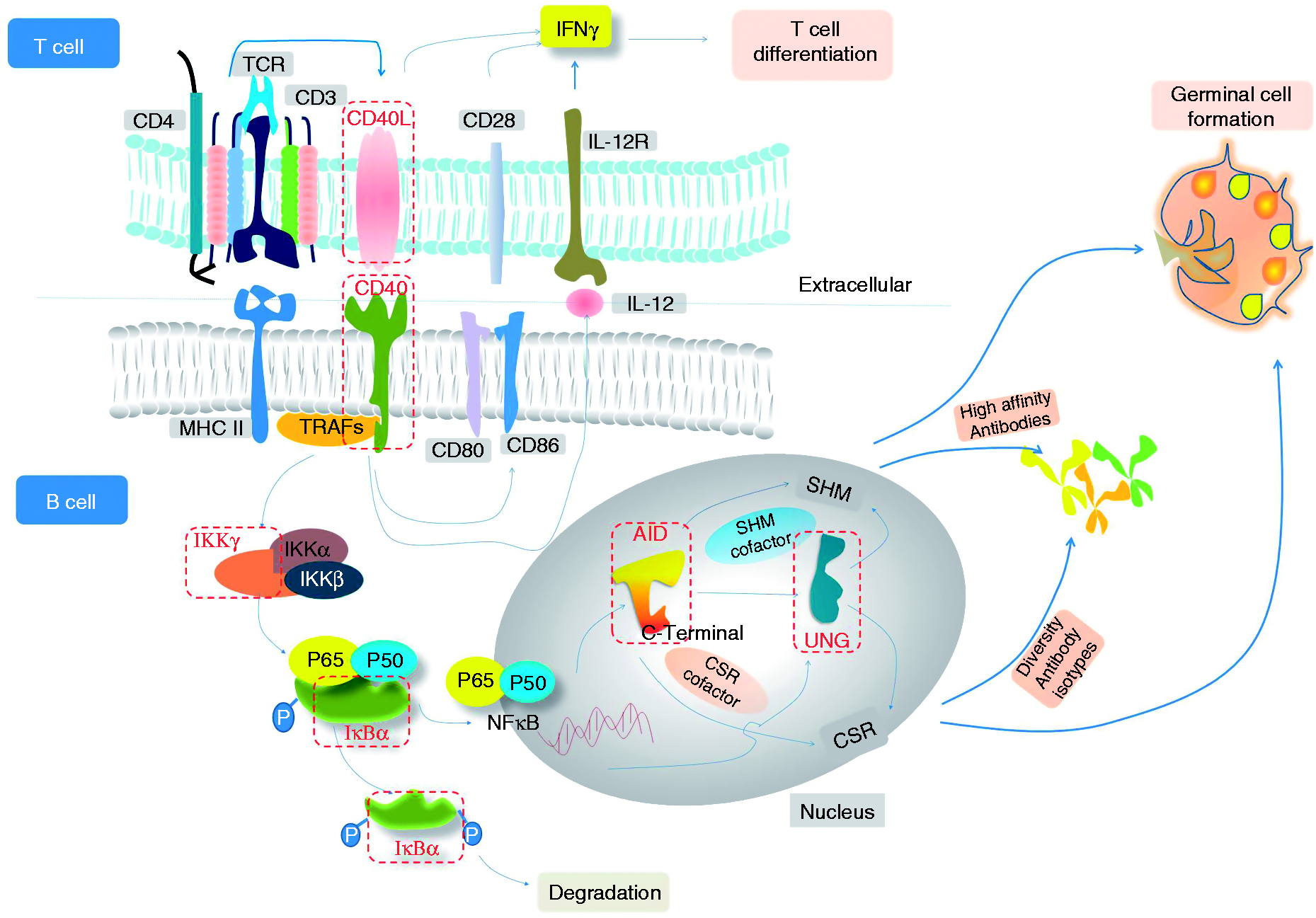
As the products of AID and UNG are expressed in B cells, HIGM2, HIGM-AID and HIGM5 syndromes are considered as primary B-cell defects. HIGM2 and HIGM5 are characterized by lymph node hyperplasia with giant GCs and defective CSR and SHM. However, HIGM-AID is defective in the AID C-terminal, resulting in abnormal CSR with normal SHM.18,19
The defects in the NF-κB essential modulator (NEMO, IKK-γ) and functional mutations of IKBα are part of the NF-κB inhibitory complex, which results in the translocation of NF-κB into the nucleus. 20 Consequently, the expression of AID and UNG is abnormal and patients display the HIGM syndrome; these patients are classified as HIGM6. Because of the involvement of NF-κB in multiple signaling pathways, the clinical phenotype is more severe than other gene deficiencies.21–27
Other HIGM syndromes not linked to the defects of CD154, NEMO, AID, CD40 or UNG have also been reported, such as genetic deficiencies in PMS2, 28 ATM, 29 and NBS1. 30 These syndromes are termed HIGM4,28–30 and they all have a defect in the CD40/CD40L signaling pathway in B cells leading to the failure of CSR or SHM, even though they had normal CD40L. This form clinically resembles HIGM2, including lymphoid tissue hyperplasia, but without GC formation. CSR is impaired, whereas SHM is normal. 31
Clinical manifestations
The clinical manifestations of HIGM are very complicated owing to its complex pathogenesis. Blockade in the CD40L and CD40 interaction results in opportunistic infections. Most patients with HIGM1 and HIGM3 syndromes present clinical manifestations during their first or second year after birth. 32 An opportunistic infection of Pneumocystis jiroveci (carinii) pneumonia is most common during the first year of life in some patients (an approximately 40% incidence), accompanied by respiratory tract infections.7,33,34 Other opportunistic infections have also been reported, such as infections with Cytomegalovirus, Cryptococcus, Candida, Bartonella and hepatitis virus.7,33,34 Gastrointestinal complaints with diarrhea and malabsorption are the second most frequent occurrence in patients with XHIGM and CD40 deficiency. Gastrointestinal symptoms may be caused by a Cryptosporidium infection, which may lead to sclerosing cholangitis, a severe disease of the liver.35,36 Up to 50% of patients develop neutropenia transiently or persistently.7,33,34 The pathogenesis of the neutropenia is unknown, but most patients respond to treatment with granulocyte colony-stimulating factor. Severe neutropenia was often associated with oral ulcers, proctitis and skin infections.7,33,34 Autoimmune manifestations that occur in patients with XHIGM syndrome or CD40 defects include chronic arthritis, low platelet counts (thrombocytopenia), hemolytic anemia, hypothyroidism and immune complex-mediated nephritis. 37 The risk for hepatocellular carcinoma was increased in patients with XHIGM or CD40 deficiency.35–40 A few patients with HIGM syndrome develop a rapidly progressive neuroendocrine carcinoma.35–40
The clinical manifestations of patients with defects in AID and UNG included lymph node hyperplasia caused by the presence of giant GCs. 41 AID defects included autoimmunity or inflammation, diabetes mellitus, polyarthritis, autoimmune hepatitis, hematologic abnormalities, Crohn’s disease and chronic uveitis. 37 Patients with HIGM-AID with partial mutations in the C terminal end of the AID gene had recurrent infections but none exhibited autoimmunity.7,35,42–44 Patients with a defect in NF-κB activation (HIGM6) have more severe clinical manifestations than with other types of HIGM. 21
The treatments were mainly based upon administration of therapeutic Ig, antibiotics for infections and steroids used to treat neutropenia or severe autoimmune manifestations. 40 This therapeutic regime has been effective in 70% of HIGM patients, but a majority of patients still die in the second decade of life as a consequence of impaired T-cell function.45,46 Stem-cell transplantation is an available therapeutic option for CD40/CD40L interaction defects. 47 Until now, efficient drug therapy, which can correct the defective CD40/CD40L pathway and improve humoral immunity, has only been explored and developed in an extremely limited manner. HIGM patients still require many drugs, which cannot cure or relieve the immune disorder without the high risk of death or burden of autoimmune diseases.
Clinical trial therapy based on correcting the CD40/CD40L pathway
All molecules involved in the CD40/CD40L pathway to promote SHM and CSR may be candidate drug targets for HIGM; as shown in Figure 1, the deficient molecules (labeled in red) will be key targets for HIGM therapeutics. Regretfully, there are not many clinical trials for HIGM that target these molecules.
After considering all the information of the HIGM clinical trials, regulating the expression of CD40L or the CD40L functional familiar protein was identified as the most important way to cure HIGM1 and HIGM3. Both soluble forms of CD40L that were used as a CD40 agonist and an agonistic CD40 antibody were tested in clinical trials against HIGM1 and HIGM3.27,48 In a mouse model of HIGM1, treatment with human recombinant CD40L protein protected the mice from opportunistic infections, restored the mice’s ability to make gamma globulin and improved survival. Furthermore, recombinant CD40L induced monocytes in peripheral blood mononuclear cells of XHM patients in vitro to produce IL-12, as well as to up-regulate the expression of CD80/86, which, in turn, allowed their T cells to produce TNF-α and IFN-γ. Three children with HIGM1 were administered recombinant CD40L. The specific Ab responses to T cell-dependent Ags was still absent; however, both the CD4+ and CD8+ T cells in the patients developed a new capacity to respond to T-cell mitogens with the synthesis of TNF-α and IFN-γ. Additionally, on biopsy CD40L therapy improved the size and structure of the lymph nodes. This clinical study showed that recombinant CD40L is capable of improving T cell immune function in HIGM1 patients. 49 Xiying Fan et al. tested CP-870893, a human CD40 agonist monoclonal Ab, in the treatment of two HIGM1 patients with biliary cryptosporidiosis. 27 CP-870893 activated the B cells and APCs in vitro, restoring class switch recombination in patients’ B cells and inducing cytokine secretion by monocytes. Compared with recombinant CD40L, CP-870893 was more efficient in up-regulating the secretion of pro-inflammatory cytokines IL-12 and TNF-α, coupled with a lower level of production of IgE in patients’ B cells. CP-870893 infusions were well tolerated and showed significant activity in vivo, decreasing leukocyte concentration in peripheral blood, but without specific Ab responses. 48
The CD40 agonists and an agonistic CD40 Ab failed to restore specific Ab synthesis in the B cells. The incomplete activation of the CD40 receptor regulated by CD40 agonists may lead to failure in high-affinity Ab synthesis.
The mechanism of interaction between CD40 and CD40L is still not clear, especially for the structural changes that occur during pathway activation. Even though Ab synthesis recovered partially in a mouse model, this may not predict the outcome in humans. Therefore, a full evaluation of therapy with CD40 agonists is important for HIGM therapy even if CD40 agonist does not correct the immunodeficiency. CD40 agonists therapy may be prone to suppress the infection associated with T-cell function if the patients are also treated with hematopoietic stem cell transplantation (HSCT). 35 As a result, a better understanding of CD40/CD40L co-stimulation pathways and the natural functional conformation of CD40L will play important roles in modifying CD40 agonists and improving the clinical observation of HIGM.
Gene therapy was supposed to be the fundamental therapeutic solution for HIGM; however, gene therapy for HIGM still remains at the animal testing stage due to the complicated pathogenesis and the widespread involvement of CD40/CD40L in various signaling pathways. 50 Since there has been successful gene therapy for other inherited rare diseases and a gene therapy clinical trial for cancer modulated CD40L expression, these cases inspire the possibility of HIGM cure; however, attempts to transfer the CD40L gene by a recombinant retrovirus in CD40L-deficient mice to correct humoral immunity resulted in aberrant CD40L expression51–53 and thymic lymph proliferative disease. It was presumed that correcting the mutant gene and regulating CD40L to the normal level might be the key solution. 54 Therefore, the experiments that corrected the CD40L gene efficiently and on target did restore the normal expression of CD40L and CD40–murine IgG Fc fusion protein (CD40-muIg) binding, and rescued IgG class switching of naive B cells in vitro. 55 More experimental data are needed, as is a full understanding of pathogenesis mechanisms. Meanwhile, a new gene therapy method, CRISPR/Cas9, may provide more promising opportunities to accurately replace the defective genes to cure HIGM.
There has been no clinical trial for the treatment of AS-HIGM. The results of attempts to target proteins, such as mutant macromolecules (AID, UNG, NEMO and IKB-α) involved in the CD40/CD40L pathway, were disappointing. Thus, we do not know if modulating these proteins may be effective against HIGM. Consequently, successfully developing HIGM therapies that target the CD40/CD40L pathway with an acceptable therapeutic index will depend on a deeper understanding of the complex biology of CD40.
Possible side effects of modulating the CD40/CD40L pathway
CD40 is the most appreciated critical regulator of cellular and humoral immunity via its expression on B lymphocytes, dendritic cells and monocytes. 56 CD40 is also expressed on the cell surface of many other normal cells, including endothelial cells, fibroblasts, hematopoietic progenitors, platelets and basal epithelial cells; the widespread physiologic effect of the CD40 signaling pathway is profound.56–58 Atherosclerosis, graft rejection, coagulation, infection control and autoimmunity are all regulated by the CD40/CD40L interactions. 56 Like some other members of the TNF receptor family, CD40 signaling is mediated, in large part, by an intricate series of downstream adapter molecules. As a consequence of CD40 signaling, a number of well-characterized signal transduction pathways are activated, including the NF-κB, MAPK and PI3K pathways. 59 These pathways are related to the cell-signaling cascade. However, a full understanding of the CD40 circuitry at the systems biology level remains incomplete. The abnormal expression of CD40L in mice has been demonstrated to induce autoAb production in vivo and also correlate with lupus-like disease or other autoimmune diseases. 60 Based on all of the clinical information to date, the best approach to correcting the defects in the CD40L/CD40 pathway in the HIGM syndrome is not clear.
Conclusion
The detection of abnormal genes in the early stages of life is crucially important for clinical therapeutic interventions, and may prevent patients from experiencing irreversible damage, as well improving their quality of life. As for inherited rare diseases, gene therapy and stem cell transplantation may provide cures. However, implementation of genetic therapeutic approaches for HIGM are premature owing, in part, to an insufficient understanding of the regulation of CD40L gene expression. Clinical trials have demonstrated that HSCT was more likely to be successful if the donor was a sibling. Although IgG replacement may improve the course for all types of HIGM, it is an inadequate long-term solution. Future research should be directed to the development of drugs that restore CD40/CD40 interactions. The recovery of specific Ab synthesis should be the most important criterion for evaluating the success of an intervention that modulates the CD40/CD40L pathway. Increased secretion of proinflammatory cytokines that can upregulate the immune system may be another sign of recovery of specific Ab synthesis. Ultimately, the ideal therapeutic regimen for HIGM may combine different therapeutic approaches with the goal of improving the entire immune system.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Guangdong Province special plan of introducing innovative R&D team (201101Y0104990178, China).