
Abstract
Skin barrier defects play an important role in atopic dermatitis (AD) pathogenesis. Loricrin, an important barrier protein suppressed in human AD, is down-regulated by IL-4 in keratinocytes. However, the molecular mechanism is unknown. Since loricrin transcription requires p300/CBP, and Stat6 also recruits this common coactivator for its stimulated factors, we hypothesize that IL-4-activated Stat6 competes for the available endogenous p300/CBP, leading to loricrin transcription inhibition. First, we showed that loricrin is suppressed in the skin of IL-4 transgenic mice, an AD mouse model. In human keratinocytes, IL-4 down-regulation of loricrin is abrogated by a pan-Jak inhibitor, suggesting that the Jak-Stat pathway is involved. To further investigate the downstream molecular mechanism, we transfected HaCat cells with a loricrin promoter and then treated them with either IL-4 or vehicle. Not surprisingly, IL-4 greatly suppressed the promoter activity. Interestingly, this suppression was prevented when we knocked down Stat6, indicating that Stat6 participates in IL-4 regulation of loricrin. A Stat6-specific inhibitor confirmed the knockdown study. Finally, IL-4 suppression of loricrin was reversed with transfection of a CBP expression vector in a dose-dependent manner. Taken together, for the first time, we delineate a molecular mechanism for IL-4 down-regulation of loricin expression in human keratinocytes, which may play an important role in AD pathogenesis.
Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disease. 1 Although the pathogenesis and etiology are not fully understood, it is generally accepted that dysregulation of immune response and skin barrier defects play an important role in the development and progression of AD.1,2 Filaggrin, involucrin and loricrin are important barrier proteins, and their roles in AD have been extensively investigated.3–6 In addition to polymorphisms,7,8 barrier defects may also be caused by the dysregulated immune response.4,6,9 Th2 cytokines, including IL-4, are up-regulated in AD.10,11 We and others have shown that the important role of IL-4 in AD involves not only the up-regulation of epidermal chemotactic, angiogenic and pro-inflammatory genes, but also down-regulation of barrier proteins.10,12,13 Recently, the FDA approved dupilumab, a monoclonal Ab against IL-4 receptor α, for AD in the USA. 14 Loricrin, an important barrier protein suppressed in patients with AD, was reported to be down-regulated by IL-4 in primary keratinocytes, 6 but the molecular mechanism has not been delineated.
A major pathway for IL-4 signaling in keratinocytes is the Jaks-Stat6 pathway, in which activated Stat6 binds to the cis-acting IFN-γ activation site (GAS) of its target genes to regulate transcription.15–17 Stats usually recruit the coactivator p300/CBP in the general transcription machinery.18,19 On the other hand, locrin transcription also involves p300/CBP among other transcription factors and coactivators such as AP-1 and Sp1.20,21 However, Stat6 has not been reported to bind directly to the loricrin promoter, probably owing to lack of a typical Stat6 site. Based on these previous findings, we hypothesized that Stat6, activated by IL-4, may compete with the loricrin transcription complex for limited levels of the common coactivator, p300/CBP, sequestering it from the complex and thus indirectly suppressing loricrin transcription (Figure 1).
Proposed mechanism for IL-4 suppression of loricrin transcription in keratinocytes. Human loricrin transcription involves AP-1, Sp1 and the coactivator p300/CBP among other transcription factors. With IL-4 treatment, activated Stat6 translocates to the nucleus to bind to the GAS site of its target genes, where it recruits p300/CBP, thus sequestering p300/CBP from loricrin transcription complex and suppressing transcription of the human loricrin gene.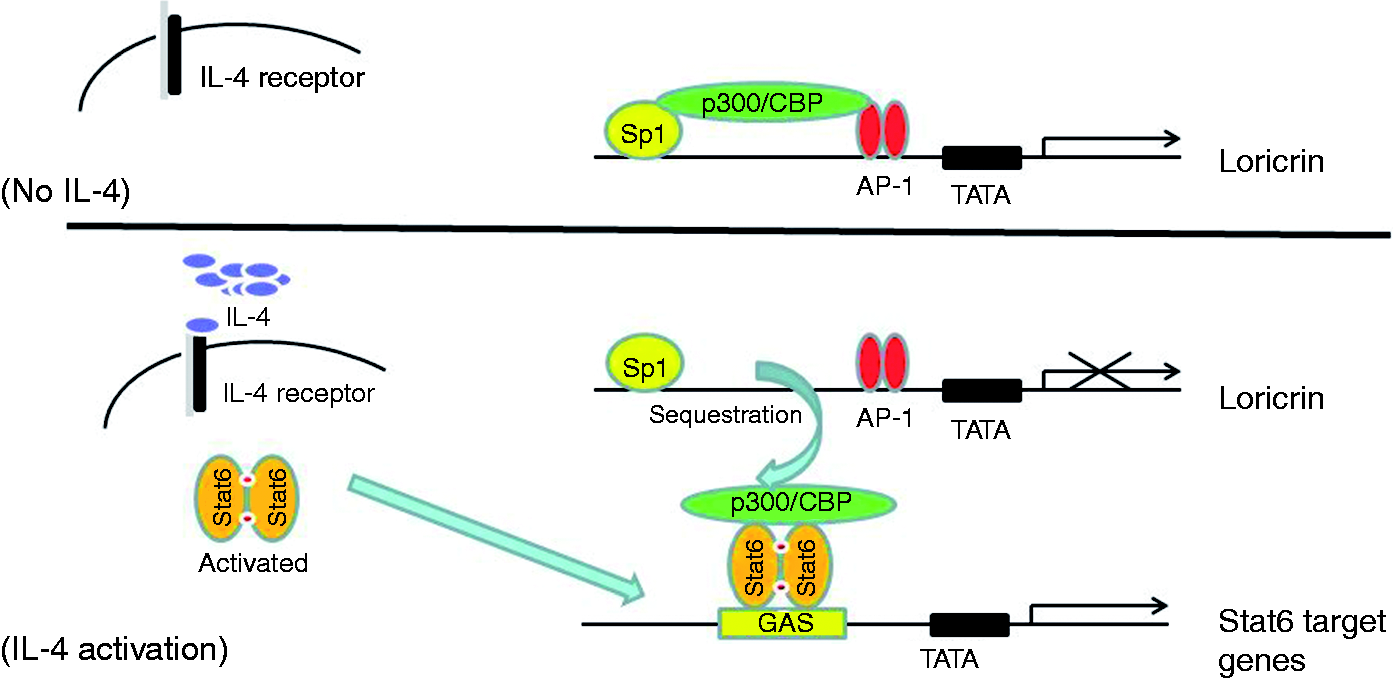
In this study, using an AD animal model and molecular biology techniques, we have described a molecular mechanism for IL-4 inhibition of loricrin transcription in human keratinocytes, which may help us further understand the effect of IL-4 in AD pathogenesis.
Materials and methods
Materials
Human recombinant IL-4, FBS, charcoal-stripped FBS, DMEM, penicillin/streptomycin, RT and Trizol reagent were purchased from Invitrogen (Carlsbad, CA, USA); Jak Inhibitor I was obtained from Calbiochem (La Jolla, CA, USA); AS 1517499 were purchased from Axon Medchem (Reston, VA, USA); SYBR Green PCR Master Mix and Attractene Transfection Reagent were purchased from QIAGEN Inc. (Valencia, CA, USA); anti-loricrin Ab was purchased from Abcam (Cambridge, MA, USA); anti-GAPDH Ab was from Novus Biologicals (Littleton, CO, USA); Passive Lysis Buffer and Dual-Luciferase Reporter Assay were from Promega (Madison, WI, USA); the DN-Stat6, DN-Stat3 and CBP expression vector were kindly provided by Dr. Steven M Dubinett (University of California at Los Angeles, Los Angeles, CA, USA), 22 Dr. Toshio Hirano (Osaka University, Osaka, Japan) 23 and Dr. Richard H Goodman (Oregon Health & Science University, Portland, OR, USA), 24 respectively.
Mice
The IL-4 Tg mice were kept at 25℃ in a pathogen-free environment. They develop AD-like skin lesions spontaneously as described previously. 25 The mice were sacrificed and skin tissues on the earlobes were collected before the onset of skin lesions on d 75. Wild type mice were used as controls. The tissues were put into RNAlater solution for mRNA analysis or flash-frozen for Western analysis. All experimental procedures were performed in accordance with the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee of the University of Illinois at Chicago.
Cell culture
Primary human keratinocytes were cultured in EpiLife Medium plus Keratinocyte Growth Supplement (Invitrogen). Then the cells were transferred to DMEM supplemented with 10% FBS for 3 d to differentiate (high calcium). HaCat cells (immortalized human keratinocytes) were grown in DMEM supplemented with FBS (10%), non-essential amino acids and penicillin/streptomycin. The cells were incubated in a humidified atmosphere of 5% CO2 at 37℃. Fresh culture media were replaced every 48 h. Cells were treated with IL-4 in DMEM supplemented with 1% charcoal-dextran-treated FBS for 24 h. Finally, cells were washed twice with ice-cold PBS and were stored at −80℃ until assays were performed.
RNA isolation and real-time RT-PCR analysis
Total RNA from cell culture and mouse skin was isolated using Trizol reagent according to the manufacturer’s instructions. 26 The RT and real-time PCRs were performed as previously described. 27 The mouse loricrin primers used were 5′- GGA GGG GGC TAT TAC TCC TC -3′ and 5′- CAC CTC CAC AGC TAC CAC CT -3′; the mouse GAPDH primers used were 5′-AGA ACA TCA TCC CTG CAT CC-3′ and 5′-ACA CAT TGG GGG TAG GAA CA-3′; the human loricrin primers used were 5′-AGT GGA CTG CGT GAA GAC CT -3′ and 5′-AGC CAG AAC CGC TGC TAC -3′; the human GAPDH primers used were 5′-ACA CCC ACT CCT CCA CCT TT-3′ and 5′-TGC TGT AGC CAA ATT CGT TG-3′. The PCR was carried out in duplicate on a Stratagene Mx3000 real-time PCR machine (Santa Clara, CA, USA).
Western blot analysis
Western blots were performed as described previously. 4 Briefly, cellular proteins were separated on a 4–12% SDS-PAGE gel under reducing condition and transferred to a PVDF membrane. The membrane was blocked by Li-Cor blocking buffer (Li-Cor, Lincoln, NE, USA) for 1 h at room temperature (23℃) and then incubated with the primary Ab overnight (16 h) at 4℃ on a rocking platform. After a series of washes, membranes were incubated with Li-Cor secondary Abs linked to infrared dyes (IRDye 680 or IRDye 800) for 1 h at room temperature. Finally, the blots were scanned by a Li-Cor Odyssey machine and quantified by the software ImageJ (NIH, Bethesda, MD, USA).
Generation of the loricrin promoter–firefly luciferase reporter vector
Oligonucleotides were synthesized and PAGE purified (IDT, Coralville, IA, USA). The sense strand was 5′- CTT CAT GAA TGG GCC ATC CTG TGC CTG CAA TCA CAG GGA GGT GGG GCC GAC AGC CAC GGG TCA CGT AAC TGA GGC CAA ACA CAA GAA GCT GGC CTG GAT CAA TGA GTC AGG GAG AGC TCT ATA TAT AAC CTC AGG AGA TCA GTG CTC CTC ACA TTG CCA -3′, and the antisense strand was 5′- AGC TTG GCA ATG TGA GGA GCA CTG ATC TCC TGA GGT TAT ATA TAG AGC TCT CCC TGA CTC ATT GAT CCA GGC CAG CTT CTT GTG TTT GGC CTC AGT TAC GTG ACC CGT GGC TGT CGG CCC CAC CTC CCT GTG ATT GCA GGC ACA GGA TGG CCC ATT CAT GAA GGT AC -3′. This piece of the loricrin promoter (-159 to +1) is essential and adequate for loricrin transcription.20,21 We subcloned the promoter into the KpnI and HindIII sites of pGL3-Enhancer Vector (Promega).
Transfection of HaCat cells
Attractene transfection was performed according to the manufacturer's instructions. Briefly, HaCat cells were seeded in 12-well plates and cultured until 60% confluency. An equal amount of DNA was transfected per well. Eighteen h after transfection, the cells were treated with IL-4 (100 ng/ml) for another 24 h. Transient expression of the reporter gene was quantified by a standard luciferase assay and was normalized against renilla luciferase. Luciferase activities were measured using a luminometer (Glomax 20/20 luminometer; Promega)
Statistical analysis
Data were examined by t-test (Figures 2 and 3), one-way ANOVA followed by the Tukey’s test (Figures 4, 6 and 7) or two-way ANOVA followed by Bonferroni post-tests (Figure 5) using Prism software (GraphPad Software, San Diego, CA, USA). Values were considered statistically significant at P < 0.05.
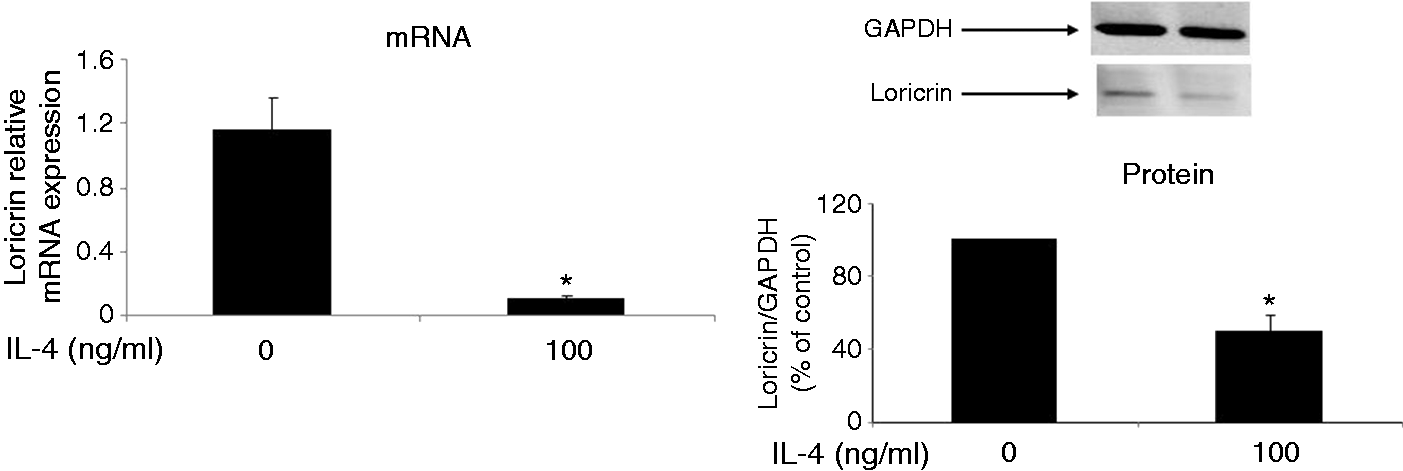
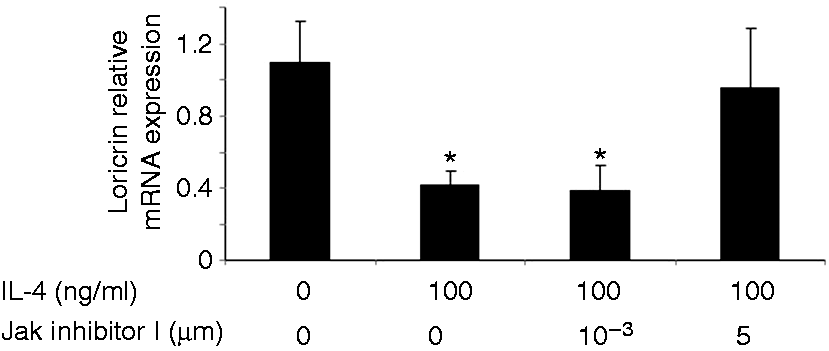
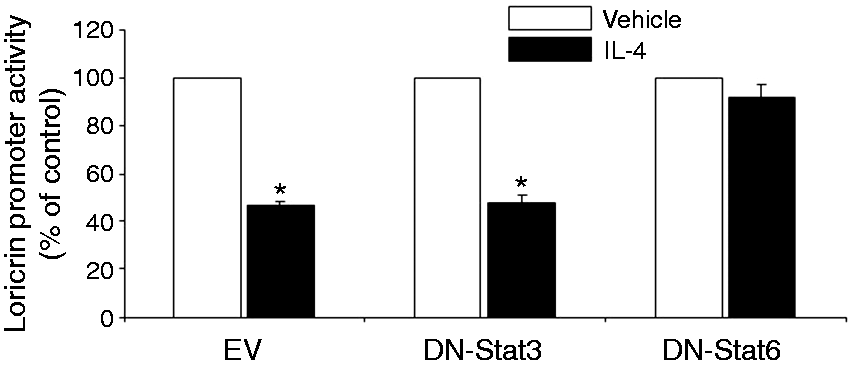
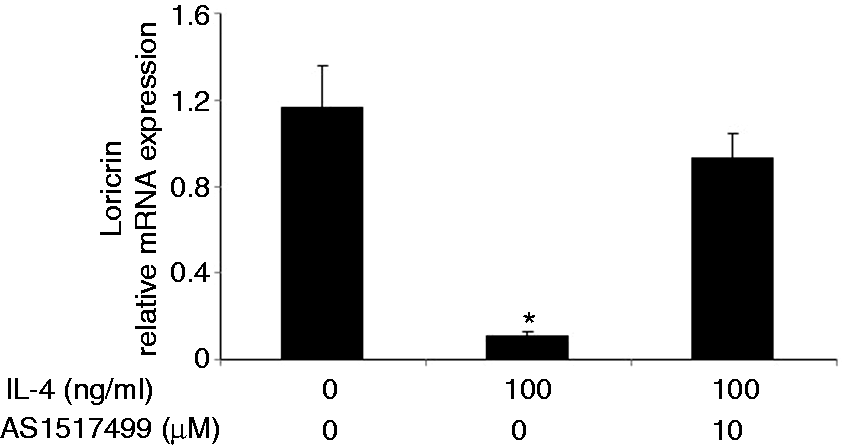
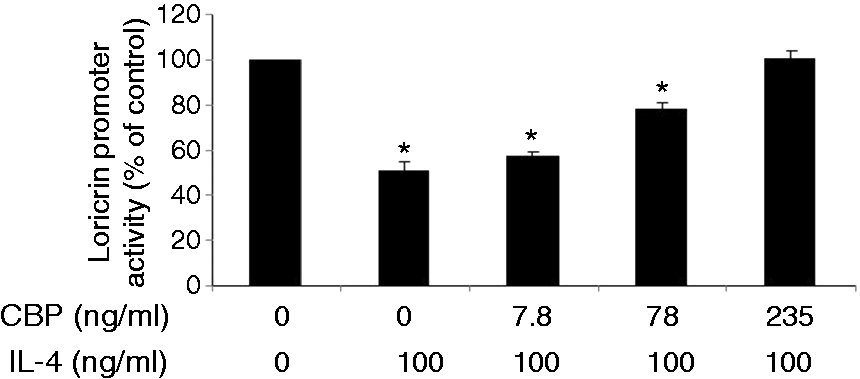
Loricrin expression is down-regulated in the non-lesional skin of IL-4 Tg mice. The expression of loricrin was compared between wild type (WT) mice and IL-4 transgenic (Tg) mice at both mRNA and protein levels. GAPDH were used as the internal reference. Skin tissues were collected before the onset of skin lesions. Values are expressed as the mean ± SEM (n = 3). *P < 0.05 vs. wild type mice. IL-4 down-regulates the expression of loricrin in primary human keratinocytes. Primary human keratinocytes (PHK) were treated with IL-4 for 24 h. Loricrin expression was analyzed by real-time RT-PCR and Western blot. GAPDH were used as the internal reference. Values are expressed as the mean ± SEM (n = 3). *P < 0.05 vs. 0 ng/ml of IL-4. Jak inhibitor I abolishes IL-4 down-regulation of loricrin expression in primary human keratinocytes. Total RNA obtained from PHKs treated with Jak inhibitor I (a pan Jak inhibitor) was subjected to real-time RT-PCR analysis. GAPDH were used as the internal reference. Values are expressed as the mean ± SEM (n = 3). *P <0.05 vs. control. Dominant negative Stat6 prevents the suppression of loricrin promoter activity by IL-4 in HaCat cells. HaCat cells were co-transfected with the loricrin promoter reporter construct, a renilla luciferase plasmid, and with empty vector, dominant negative (DN)-Stat3 or DN-Stat6 for 18 h. Cells were then treated with either IL-4 or vehicle for another 24 h. The experiment was repeated three times with triplicate wells for each group. Values are expressed as the mean ± SEM. *P < 0.05 vs. vehicle. AS 1517499 abolishes IL-4 down-regulation of loricrin expression in primary human keratinocytes. PHKs were treated with IL-4 and AS 1517499 (a Stat6-specific inhibitor) for 24 h and were subjected to real-time RT-PCR analysis. GAPDH were used as the internal reference. Values are expressed as the mean ± SEM (n = 4). *P <0.05 vs. control. CBP expression vector abrogates the suppression of loricrin promoter activity by IL-4 in HaCat cells. HaCat cells were co-transfected with the loricrin promoter reporter construct, the renilla luciferase plasmid, and with either empty vector (0 ng/ml of CBP) or increasing doses of a CBP expression vector for 18 h. Equal amounts of DNA were transfected in each group. Cells were then treated with either IL-4 or vehicle for another 24 h. The experiment was repeated three times with triplicate wells for each group. Values are expressed as the mean ± SEM. *P < 0.05 vs. control.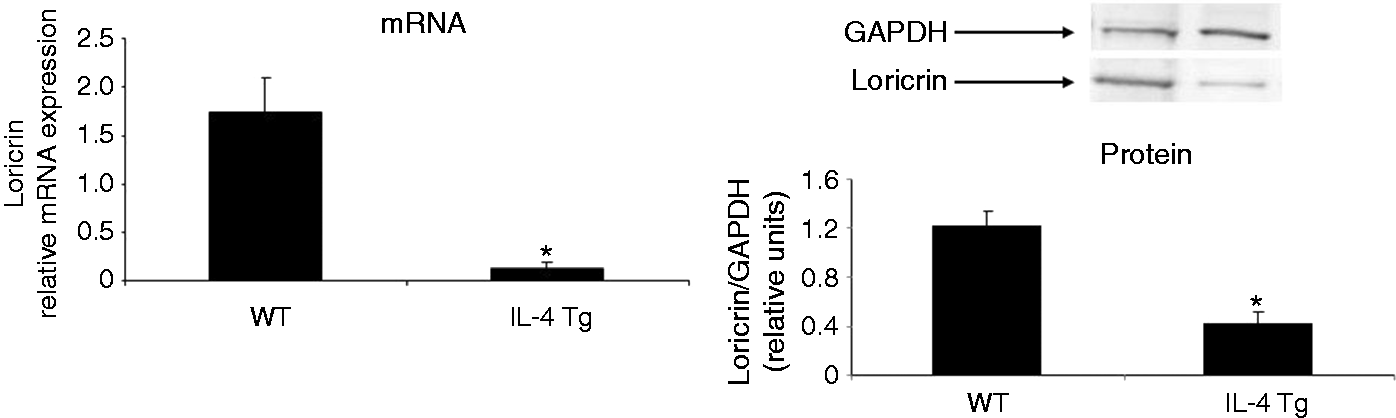
Results
Loricrin is suppressed in IL-4 Tg mice
We have generated an AD animal model, IL-4 transgenic (Tg) mice, by overexpressing IL-4 in the basal epidermis. 25 These mice spontaneously develop AD-like skin lesions, 25 which meet the clinical and histological diagnostic criteria for human AD. The pruritic skin lesions were evidenced by constant face rubbing and repeated scratching in IL-4 Tg mice. 25 The skin lesions initially develop on the earlobes and subsequently extended to other parts of the body. Scratching may cause infection leading to pustules and crusting. 25 We collected skin tissues from the IL-4 Tg mice before the onset of skin lesions to exclude the interference from skin inflammation. Wild type mice were used as controls. We demonstrated that loricrin is suppressed at both mRNA and protein levels, which is consistent with clinical AD findings (Figure 2).
Jaks are involved in IL-4 down-regulation of loricrin expression in keratinocytes
To further examine whether the down-regulation of loricrin expression in IL-4 Tg mice is due to direct action of IL-4, we treated primary human keratinocytes with IL-4, and found that IL-4 treatment indeed suppressed loricrin expression at both the mRNA and protein levels (Figure 3). Since the Jaks-Stat6 pathway is a major pathway for IL-4 signaling in keratinocytes, we examined whether inhibition of Jaks may prevent IL-4 down-regulation of loricrin mRNA expression. We treated primary keratinocytes with IL-4 and increasing doses of Jak inhibitor I, a pan-Jak inhibitor. Jak inhibitor I abolished IL-4 suppression of loricrin transcription in keratinocytes at 5 µM (Figure 4).
Stat6 is involved in IL-4 down-regulation of loricrin expression in keratinocytes
We generated a loricrin–firefly luciferase construct and proposed to determine whether knockdown of individual Stats may affect IL-4 action on loricrin promoter activity. Since primary human keratinocytes are refractory to transfection, we used HaCat cells, a human keratinocyte cell line. We co-transfected HaCat cells with the loricrin promoter construct and with an empty vector, dominant negative (DN)-Stat3 or DN-Stat6, which have been used successfully to specifically knock down Stat3 or Stat6 by us and others. Renilla luciferase construct was used as internal reference. Our data show that Stat6 but not Stat3 is functionally involved in IL-4 suppression of loricrin promoter activity (Figure 5), although the phosphorylation of both Stat3 and Stat6 is stimulated by IL-4 in keratinocytes. 15 To further confirm this conclusion, we used a Stat6-specific inhibitor, AS 1517499. As shown in Figure 6, this inhibitor abolished IL-4 down-regulation of loricrin mRNA expression. Taken together, our data indicate that IL-4 signals through the Jaks-Stat6 pathway to down-regulate loricrin expression in keratinocytes.
IL-4 inhibition of loricrin transcription may be due to Stat6 competitive sequestration of CBP in keratinocytes
It is known that p300/CBP is very important for loricrin transcription. 20 Previously we have demonstrated physical interaction between Stat6 and CBP in keratinocytes. 4 In this experiment, we co-transfected HaCat cells with the loricrin promoter and increasing doses of a CBP expression vector. As shown in Figure 7, IL-4 suppresses loricrin promoter activity without the CBP construct, but the inhibitory effect was reversed gradually with increasing doses of CBP, pointing to a competitive process.
Discussion
AD is a chronic inflammatory skin disease affecting many individuals in developed countries. Epidermal barrier defects play an essential role in AD pathogenesis by causing allergen penetration, water loss and infection.1,28 In AD, elevation of Th2 cytokines may dysregulate barrier proteins.6,9,29–31 In this study, we focused on the molecular mechanism for IL-4 inhibition of loricrin transcription. Jang et al. have reported that loricrin transcription in human keratinocytes is regulated by a complex interplay among transcription factors Sp1, CREB, AP-1, AP-2 and the coactivator p300/CBP.20,21 c-Fos/c-Jun, Jun D, Sp1 and p300/CBP form the general transcription machinery to transactivate the human loricrin gene. 20 They further demonstrated that transfection of p300/CPB expression vectors stimulates loricrin transcription. 20 It is well established that p300/CBP is an adaptor molecule in the transcription complex that can acetylate histones and transcription factors.32,33 Acetylation of lysine residues on the amino-terminus of histones causes conformational changes of the nucleosome, increasing accessibility of the transcription complex to its target DNA. 34
We have shown that IL-4 regulates hundreds of genes in keratinocytes.12,13,15,16 IL-4-activated Stat6 binds to the GAS site to regulate its target genes,15,16 and a typical GAS site for Stat6 consists of two palindromic half sites TTC and GAA with four spacing nucleotides in between. 15 Probably because of a lack of typical Stat6 binding sites, Stat6 has never been reported to bind to the loricrin promoter. How Stat6 inhibits loricrin transcription without binding to its promoter area is an interesting question that we need to address . Previously we showed that IL-4 up-regulated CCL26 expression through Stat6 binding to the GAS site of the CCL26 promoter in keratinocytes. 15 Interestingly, Lim et al. demonstrated that transfection of CBP in human esophageal epithelial cells stimulates CCL26 transcription, whereas knockdown of CBP suppresses its transcription. Recently we demonstrated a physical interaction between CPB and Stat6 in keratinocytes, 4 and the carboxyl-terminal region of Stat6 is required for association with p300/CBP. 35 Since p300/CBP is the common coactivator for both loricrin and Stat6 target genes (e.g. CCL26 and IL-1915,16), IL-4 signaling in keratinocytes may lead to activated Stat6 recruiting p300/CBP, thus sequestering this coactivator from the loricrin general transcription machinery (Figure 1).
IL-4 has been shown to signal through different pathways, such as Jak/Stat, NF-κB, MAPK and PI3.36,37 Our data show that IL-4 inhibition of loricrin is at the transcriptional level through the Jaks-Stat6 pathway. Interestingly, our preliminary data suggest that IL-4 may also down-regulate loricrin and other barrier proteins post-transcriptionally through regulating microRNAs. Further studies are required to fully characterize the molecular mechanisms involved.
Th2 cytokines, including IL-4, IL-5 and IL-13, play an important role in AD pathogenesis. In addition, Th1, Th17 and other pro-inflammatory cytokines are also involved in AD pathophysiology.1,38 Interestingly, Yagi et al. showed that AD-like skin lesions occur in Stat6-deficient NC/Nga mice. 39 Furthermore, as compared with common classical extrinsic AD, intrinsic AD (15–20%) is characterized by higher levels of IFN-γ and lower expression of Th2 cytokines. 11
Taken together, results of this investigation delineate, for the first time, a competitive mechanism by which IL-4 inhibits loricrin transcription by sequestration of CBP in keratinocytes. This molecular mechanism may provide an explanation for loricrin suppression in AD. The data of this study, together with our previous data showing induction of AD by transgenic IL-4 expression and other skin barrier protein suppression by IL-4, provide further support for the notion that immune mechanisms are capable of not only triggering AD development, but also maintaining a feed-forward mechanism of inflammation by suppressing barrier proteins, thereby allowing penetration of allergens that stimulate the inflammatory process.
Footnotes
Acknowledgments
We would like to thank Dr. Steven M Dubinett for the DN-Stat6, Dr. Toshio Hirano for the DN-Stat3 and Dr. Richard H Goodman for the CBP expression vector.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was, in part, supported by the Albert H. and Mary Jane Slepyan Endowed Fellowship Fund (L.B.) and the Dr. Orville J. Stone Endowed Professorship in Dermatology Fund (L.S.C.).