
Abstract
Alveolar macrophages (AMs) are multitasking cells that maintain lung homeostasis by clearing apoptotic cells (efferocytosis) and performing antimicrobial effector functions. Different PRRs have been described to be involved in the binding and capture of non-opsonized Streptococcus pneumoniae, such as TLR-2, mannose receptor (MR) and scavenger receptors (SRs). However, the mechanism by which the ingestion of apoptotic cells negatively influences the clearance of non-opsonized S. pneumoniae remains to be determined. In this study, we evaluated whether the prostaglandin E2 (PGE2) produced during efferocytosis by AMs inhibits the ingestion and killing of non-opsonized S. pneumoniae. Resident AMs were pre-treated with an E prostanoid (EP) receptor antagonist, inhibitors of cyclooxygenase and protein kinase A (PKA), incubated with apoptotic Jurkat T cells, and then challenged with S. pneumoniae. Efferocytosis slightly decreased the phagocytosis of S. pneumoniae but greatly inhibited bacterial killing by AMs in a manner dependent on PGE2 production, activation of the EP2–EP4/cAMP/PKA pathway and inhibition of H2O2 production. Our data suggest that the PGE2 produced by AMs during efferocytosis inhibits H2O2 production and impairs the efficient clearance non-opsonized S. pneumoniae by EP2–EP4/cAMP/PKA pathway.
Introduction
Alveolar macrophages (AMs) maintain airway homeostasis, serve as the first line of defense against microorganisms and function in the clearance of apoptotic cells (i.e. efferocytosis) during tissue injury. Previous research from our laboratory and others has shown that phagocytosis of apoptotic cells triggers the release of prostaglandin E2 (PGE2) and other anti-inflammatory mediators, such as IL-10 and TGF-β.1–3 PGE2, the major lipid mediator synthesized by macrophages during the inflammatory response, 4 is derived from cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) activities on released arachidonic acid. PGE2 functions through four different G protein-coupled E prostanoid (EP) receptors. EP1 is coupled to Gq, resulting in increased intracellular Ca2+. EP3 is coupled to Gi and leads to a reduction in cAMP, whereas EP2 and EP4 are coupled to Gs, resulting in increased intracellular cAMP. Several studies have shown that increases in cAMP account for the inhibitory effect of PGE2.5–8 An increased cAMP concentration in AMs stimulates protein kinase A/exchange protein directly activated by cAMP (PKA/EPAC), thereby inhibiting reactive oxygen species (ROS) generation and bacterial killing. 8 Moreover, inhibition of the membrane translocation of p40phox and ROS production caused by PGE2 are reversed when AMs challenged with anti-Klebsiella pneumoniae IgG are treated with an inhibitor of PKA-RII.
Phagocytosis of apoptotic cells by macrophages promotes the uncontrolled proliferation of Trypanosoma cruzi via a mechanism that is dependent on PGE2, TGF-β and polyamine production. Indeed, a previous study showed that treatment of infected animals with COX inhibitors abolishes parasitemia. 9 A different study showed that instillation of apoptotic cells in a model of endotoxin-induced pulmonary inflammation inhibited inflammatory cell recruitment in bronchoalveolar lavage fluid and the lung and decreased proinflammatory mediator production. 10 Additionally, we previously demonstrated that inhibition of FcR-mediated phagocytosis due to AM efferocytosis is mediated by the PGE2/EP2 receptor/cAMP axis. 3
Patients with asthma, chronic obstructive pulmonary disease (COPD) or who have inhaled toxic agents accumulate a large number of apoptotic cells due to a deficiency in the phagocytosis of these cells mediated primarily by AMs. Statins and corticosteroids have been used as adjuvant therapies in patients with COPD, to enhance efferocytosis.11,12 However, improvement in efferocytosis by these drugs also increases the susceptibility of mice to S. pneumoniae infection. 13
We previously demonstrated that instillation of apoptotic cells in vivo induced an increase in PGE2, bacterial load in the lung and spread of the infection to the bloodstream. 3 However, further definition of the role of non-opsonic receptors during the infection of AMs via apoptotic cells ingestion is necessary. In this study, we hypothesized that PGE2 produced during efferocytosis mediates a reduction in the effector functions of AMs infected with non-opsonized S. pneumoniae.
Materials and methods
Materials
Tryptic soy broth (TSB) and blood agar base n°2 were purchased from Acumedia (Baltimore, MD, USA). RPMI 1640 medium, FBS, indometacin, FITC, trypan blue, PBS and campothecin were obtained from Sigma-Aldrich (St. Louis, MO, USA). An EP2 agonist (Butaprost), an EP4 agonist (CAY10598) and PGE2 were obtained from Cayman Chemical (Ann Arbor, MI, USA). EP2 (AH 6809) and EP4 (AH23848) antagonists, activator of adenylyl cyclase (forskolin), inhibitor of adenylyl cyclase (SQ 22536), PKA inhibitors (KT5720, H89 dihydrochloride and PKI6-22 amide) and a PKA activator (8-Bromo-cAMP) were purchased from Tocris Bioscience (Bristol, UKP). Saponin was purchased from Acros Organic (Bridgewater, NJ, USA). Gentamycin was obtained from Life Technologies (Carlsbad, CA, USA). Annexin V and propidium iodide (PI) were purchased from BD Biosciences (San Jose, CA, USA). Cell Tracer Far Red ™ was purchased from Thermo Fisher Scientific (Waltham, MA, USA).
Animals
Female Wistar rats (∼150 g) were obtained from the Centro Multidisciplinar para Investigação Biológica – CEMIB/UNICAMP, São Paulo, Brazil. The animals were treated according to the Brazilian College of Animal Experimentation guidelines for the use of experimental animals. The institution’s animal ethics committee approved the housing conditions and experimental procedures (protocol numbers 28/2011 and 36/2016).
AMs
Resident AMs were obtained as previously described by Peters-Golden et al. 14 Briefly, AMs were obtained via bronchoalveolar lavage (BAL) and maintained in RPMI 1640 medium without FBS for 1 h. The medium was then replaced with complete RPMI 1640 with 10% FBS and 50 µg/ml gentamicin, and the cells were cultured overnight (18 h).
Generation of apoptotic cells
Jurkat cells were used as a source of apoptotic cells. These cells were incubated with camptothecin (2 µg/ml) for 5 h as previously described. 3 The percentage of early (Annexin+/PI–) and late (Annexin+/PI+) apoptotic cells was determined by flow cytometry (FACSCanto™; BD Biosciences) after labeling with Annexin V/PI, as previously described. 15
Streptococcus pneumoniae and FITC-labeled bacteria
S. pneumoniae cells (ATCC 49619) were grown in TSB at 37℃ at 200 rpm for 5 h. The estimated number of bacteria was determined using a standard curve [OD (λ = 600 nm) vs. CFU] previously established in our laboratory. The bacterial inoculum was confirmed by CFU recovery on blood agar plates. S. pneumoniae cells were conjugated with FITC as previously described. 16
Phagocytosis assay
Phagocytosis of FITC-labeled S. pneumoniae (S. pneumoniae–FITC) was assessed as previously described.7,17 Briefly, 2.5 × 105 AMs were incubated in black-wall/clear-bottom sterile 96-well microplates for 16 h in RPMI 1640 medium. The culture medium was then removed and replaced with warm, serum-free RPMI 1640 medium. Inhibitors and antagonists were added 30 min prior to the addition of apoptotic cells and/or cells incubated with the bacteria. Apoptotic cells were added to the culture in a 3:1 AC:AM ratio for 1 h as previously described, 3 followed by incubation with 100 S. pneumoniae–FITC cells:1 AM (MOI of 100) for 3 h. Trypan blue (250 µg/ml, pH 4.4) was added to the culture for 10 min to quench the fluorescence of extracellular bacteria. The fluorescence intensity was measured using a microplate fluorometer (485ex/535em, BioTek, Synergy H1). The results are expressed as the percent phagocytosis compared with a control containing S. pneumoniae + AMs (% of control).
Apoptotic Jurkat cells were stained with 5 µM Cell Tracer Far Red™ and incubated with AMs for 4 h. To evaluate efferocytosis by AMs, the percentage of intracellular stained positive cells was evaluated by flow cytometry (BD FACSCanto™).
Survival of S. pneumoniae in AMs
AMs (2 × 105 cells) were incubated overnight in 96-well microplates. The medium was then removed and replaced with warm serum-free RPMI 1640 medium. Inhibitors, agonists and antagonists were added 30 min prior to the addition of apoptotic cells and/or cells infected with S. pneumoniae. Apoptotic cells were added to the culture in a 3:1 AC:AM ratio for 1 h or 18 h as previously described, 3 followed by incubation with 50 viable S. pneumoniae cells:1 AM (MOI of 50) for 3 h for phagocytosis. The AMs were washed three times and incubated for 4 h to evaluate the survival of S. pneumoniae within the AMs. The cells were lysed with 200 μl TSB + 0.5% saponin, and serial dilutions of the cell lysate were plated onto blood agar plates. CFU recovery was evaluated after 24 h.
Quantification of NO
NO was measured indirectly by quantifying nitrite metabolites, as previously described by Griess 18 AMs were incubated with or without apoptotic cells, followed by incubation with S. pneumoniae. The culture supernatant was collected at different time points and quantified colorimetrically.
Quantification of H2O2
Intracellular levels of H2O2 were determined as previously described by Houde et al. 19 Briefly, 2.5 × 105 AMs were incubated for 30 min in 96-well microplates in the presence or absence of a PKA inhibitor (KT5720: 1 μM; H89: 10 μM; PKI6-22: 5 μM), and apoptotic cells (3:1 AC:AM ratio) were added to the culture for 1 h followed by challenge with S. pneumoniae for 30 and 90 min in the presence of 25 µM 123-DHR. The fluorescence intensity was measured using a microplate fluorometer (485ex/535em, BioTek, Synergy H1). The data are expressed as the percentage of relative changes in H2O2 production (% of AMs + Sp).
Statistical analysis
The results were analyzed using the statistical program Prism 5.0 (GraphPad Software, La Jolla, CA, USA); the data are reported as mean ± SEM. Comparisons among three or more groups were assessed with ANOVA, followed by Dunnett’s post-hoc test or two-way ANOVA followed by Bonferroni’s post-test. Comparisons between two groups were assessed using t-test analyses. Differences were considered significant if the P-values were < 0.05.
Results
PGE2/EP2/adenylyl cyclase/cAMP inhibits the phagocytosis of S. pneumoniae by AMs within the context of efferocytosis
Our group previously demonstrated that phagocytosis of apoptotic Jurkat cells by AMs induces PGE2 production and promotes the inhibition of FcR-mediated phagocytosis.
3
Here, we investigate whether efferocytosis by AMs inhibits S. pneumoniae phagocytosis, as well as the role of PGE2 in inhibiting the effector mechanisms of AMs. We evaluated AM efferocytosis using apoptotic Jurkat cells stained with Cell Tracer Far Red ™. After 4 h, ∼44% of AMs were able to phagocytose apoptotic cells (Figure 1a). AMs were also pre-treated with EP2/EP4 antagonists, PKA inhibitors and cAMP inhibitor for 30 min and then co-cultured with Far Red-labeled Jurkat apoptotic cells, to evaluate the effect of these compounds in efferocytosis. The percentage of AMs positive for apoptotic cells–FAR Red+, as well as the MIF of these cells was evaluated. We found no significant differences of efferocytosis by AMs in the presence of different compounds. Percentage of AMs positive for apoptotic cells was as follows: AMs: AMs + AC = 40.5 ± 2.8%; AMs + EP2 antagonist + AC = 48.0 ± 9.4%; AMs + EP4 antagonist + AC = 44.2 ± 0.13%; AMs + KT5720 (iPKA) = 38.4 ± 2.25%; AMs + H89 (iPKA) = 38.85 ± 5.3%; AMs + SQ (adenylyl cyclase inhibitor) = 36.0 ± 0.07%.
PGE2 suppresses S. pneumoniae phagocytosis by AMs within the context of efferocytosis via EP2/adenylyl cyclase/cAMP. (a) AMs harvested from BAL were incubated for 4 h in the presence of apoptotic Jurkat cells labeled with Far Red and evaluated by flow cytometry; the black line indicateds AMs, and the red line is AMs + AC. AMs pre-treated for 30 min in the presence of (b) COX inhibitor (5 μM, indomethacin) or PGE2 (1 μM), (c) inhibitor of adenylyl cyclase (10 μM, SQ 22536) and (d) adenylyl cyclase activator (100 μM, forskolin). AMs were incubated with a 3:1 ratio of AC:AM for 1 h and challenged with S. pneumoniae–FITC (1:100; AM:Sp). After 3 h, trypan blue (250 µg/ml) was added to the culture, and after 10 min, the fluorescence intensity was measured using a fluorometer (485ex/535em). The result from efferocytosis is expressed as a percentage of labeled cells within macrophages (representative results of two independent experiments). The other results are expressed as percent phagocytosis vs. the control containing S. pneumoniae + AMs (% of control). Representative results of three independent experiments performed in quintuples are shown. *P < 0.05 vs. AMs + Sp, #P < 0.05 vs. AMs + AC + Sp.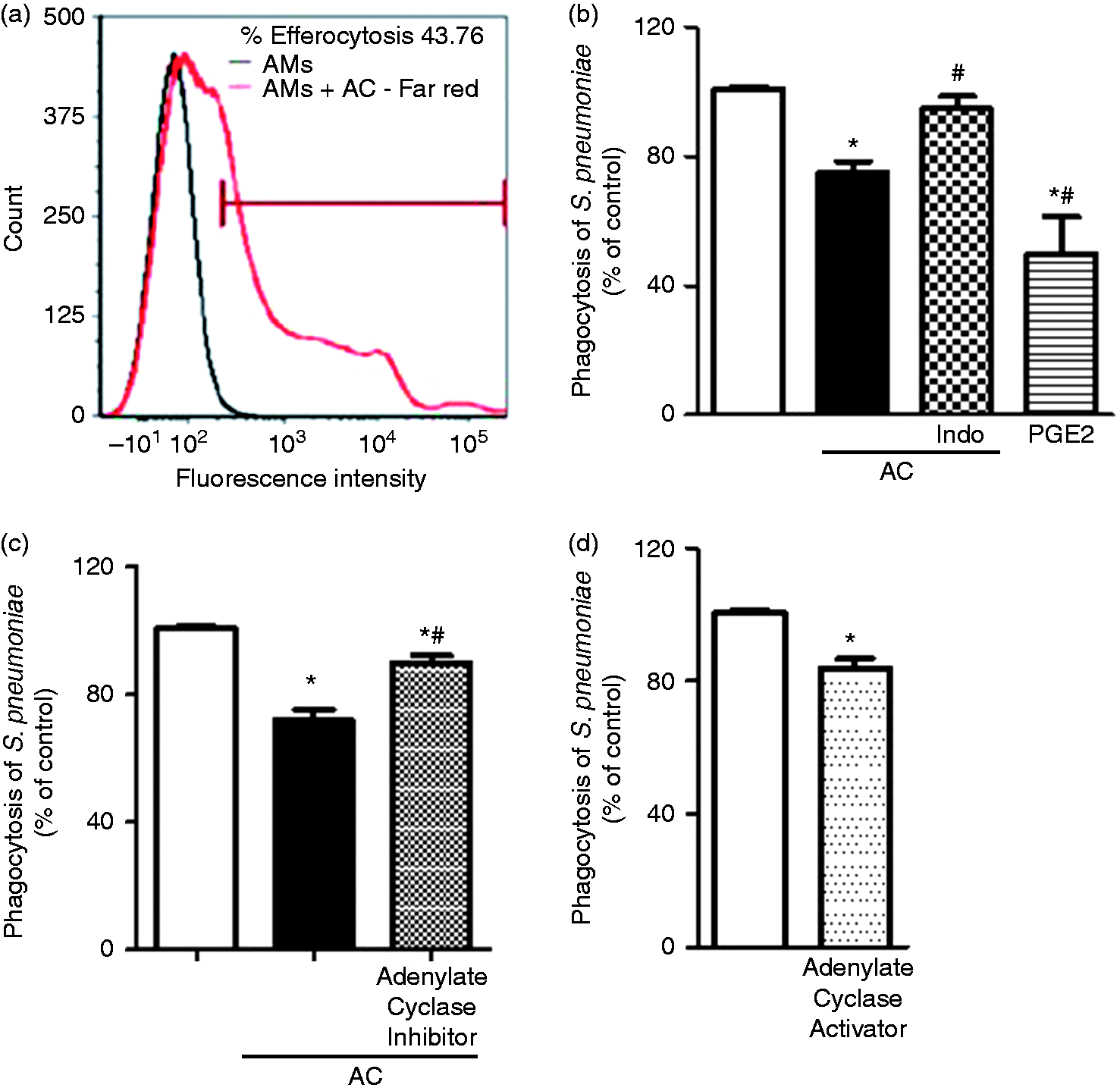
Furthermore, incubation of AMs with apoptotic cells inhibited the phagocytosis of S. pneumoniae (∼30%) compared with the control (AMs + Sp). Inhibition of S. pneumoniae phagocytosis was partially reversed by pre-treatment with the non-selective COX enzyme inhibitor indomethacin. To demonstrate that PGE2 could mimic the inhibition of S. pneumoniae phagocytosis observed during efferocytosis, AMs were pre-treated with exogenous PGE2 and then challenged with S. pneumoniae. The addition of exogenous PGE2 was more effective than apoptotic cells at inhibiting phagocytosis of the bacteria by AMs (Figure 1b).
PGE2 inhibition of macrophage effector functions has been suggested to occur through EP2 and EP4 receptor-mediated cAMP activation. To evaluate the signaling pathways by which PGE2 inhibits the effector functions of AMs containing apoptotic cells, AMs were pre-treated with the adenylyl cyclase inhibitor SQ 22536, incubated with apoptotic cells and challenged with S. pneumoniae. Adenylyl cyclase inhibition prevented the inhibitory effects of efferocytosis by AMs challenged with S. pneumoniae (Figure 1c), which was correlated with decreased phagocytosis in cells treated with the adenylate cyclase activator (forskolin) (Figure 1d). Because the phagocytosis of S. pneumoniae by AMs after efferocytosis was marginally inhibited compared with our previous observations of FcR-mediated phagocytosis inhibition (≥ 50 of inhibition), 3 we investigated the effect of efferocytosis on the antibacterial activity of AMs.
PGE2 produced during efferocytosis by AMs increases the survival of ingested S. pneumoniae
AMs were incubated in the presence of apoptotic cells for 1 h or 18 h and challenged with S. pneumoniae, and bacterial survival was then evaluated. Co-culture of AMs with apoptotic cells for 1 h or 18 h promoted the growth of S. pneumoniae in the macrophages compared with the control (AMs infected with S. pneumoniae; AMs + Sp). However, after 18 h of efferocytosis, the survival of the ingested S. pneumoniae within AMs was at least three times higher than in AMs incubated with apoptotic cells for 1 h (Figure 2a). Moreover, when AMs were initially infected with S. pneumoniae and then incubated with apoptotic cells, we observed a higher number of S. pneumoniae compared with AMs + Sp or AMs + Sp + AC (data not shown). Exposure to apoptotic cells for 18 h promotes the production of inflammatory mediators by AMs and apoptotic cells. Because the main focus of this work was to evaluate how PGE2 produced by efferocytosis could impair the effector functions of AMs against S. pneumoniae, we next evaluated the effect of apoptotic cells after 1 h of incubation with AMs.
PGE2 produced by AMs during efferocytosis suppresses the survival of ingested S. pneumoniae. (a) AMs harvested from BAL were incubated with AC for 1 h or 18 h and then challenged with S. pneumoniae (MOI of 50) for 3 h; AMs were pre-treated with (b) COX inhibitor (5 μM, indomethacin), PGE2 (1 μM), EP2 agonist (1 μM, Butaprost), EP4 agonist (1 μM, CAY10598), EP2 + EP4 agonist, (c) EP2 antagonist (10 μM, AH 6809), EP4 antagonist (10 μM, AH23848), EP2 + EP4 antagonist or vehicle for 30 min at 37℃. AMs were incubated for 1 h with a 3:1 (AC:AM) ratio or vehicle and then challenged with S. pneumoniae (MOI of 50). The AMs were lysed with TSB + saponin (0.5%) for 15 min. The lysates were plated onto blood agar for serial dilution quantification of CFU after 24 h. The results are expressed as the percent survival of ingested S. pneumoniae compared with the control containing S. pneumoniae + AMs (% of control). *P < 0.05 vs, AMs + Sp, #P < 0.05 vs. AMs + AC + Sp.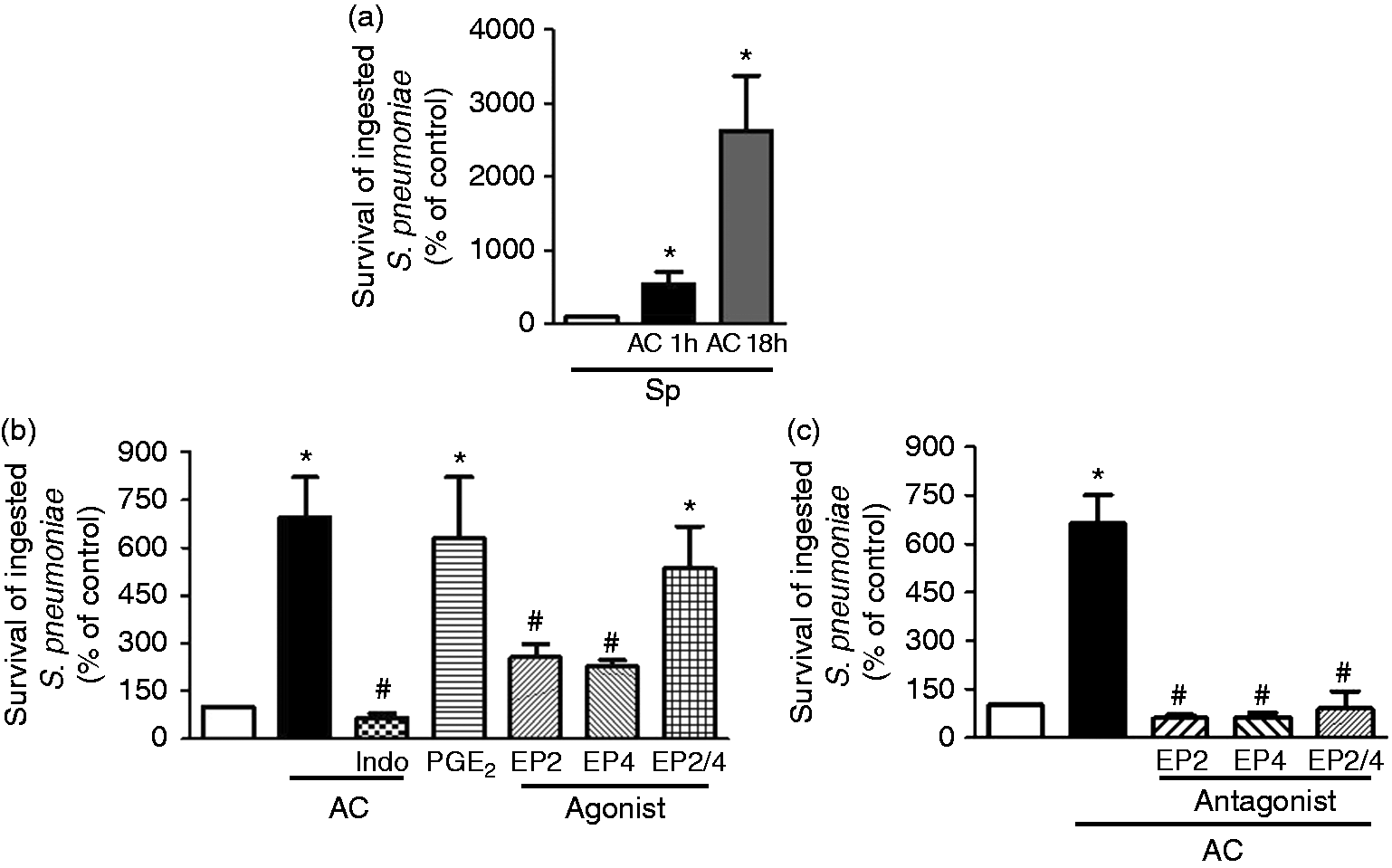
Pre-treatment with indomethacin prior to efferocytosis followed by challenge with S. pneumoniae abolished the efferocytosis-mediated inhibition of AM microbicidal activity (Figure 2b). Previous research from our laboratory and others has shown that PGE2 inhibits bacterial killing in macrophages via EP2 and EP4 receptors.3,7 To mimic the effect of PGE2 during efferocytosis, AMs were pre-treated with PGE2, and EP2 (Butaprost) and EP4 (CAY10598) agonists, and bacterial killing was then evaluated. As expected, pre-treatment with exogenous PGE2 enhanced intracellular bacterial survival. Moreover, EP2 and EP4 agonists decreased bacterial killing compared with AM + Sp. However, a synergistic effect of microbicidal activity inhibition in AMs was observed in the presence of the EP2 + EP4 agonists (Figure 2b).
Moreover, when AMs were incubated with EP2 (AH 6809) and EP4 (AH 23848) antagonists, S. pneumoniae survival was dramatically reduced compared with AMs exposed to apoptotic cells (Figure 2c). However, we did not detect any synergistic or additive effect when combinations of both antagonists were used (Figure 2c).
In addition, the effect of antagonists, agonists and inhibitors on phagocytosis and microbicidal activity was evaluated in a dose-dependent range from 10 μM to 0.1 μM (data not shown); as described in the figure legends, the lowest effective doses were used in the ensuing experiments. The concentration of antagonists, agonists and inhibitors was the same used in all microbicidal activity assays.
Efferocytosis impairs the microbicidal activity of AMs against S. pneumoniae through PGE2/cAMP/PKA
The relevant signaling cascades involved in the PGE2-mediated inhibition of bacterial killing during efferocytosis were then examined. AMs were pre-treated with SQ22536 in the presence of apoptotic cells and challenged with S. pneumoniae. Adenylyl cyclase inhibition greatly improved the microbicidal activity of AMs containing apoptotic cells (Figure 3a). To determine whether increased cAMP dampened non-opsonic microbial clearance, AMs were incubated with forskolin and challenged with S. pneumoniae. Increased cAMP production favored bacterial survival within AMs at the levels observed during efferocytosis (Figure 3b).
Efferocytosis impairs the microbicidal activity of AMs against S. pneumoniae by PGE2/cAMP/PKA. AMs harvested from BAL were pre-treated with (a) inhibitor of adenylyl cyclase (10 μM, SQ-22536), (b) activator of adenylyl cyclase (100 μM, forskolin), (c) PKA inhibitor (1 μM, PKA inhibitor KT5720 and 10 μM PKA inhibitor (H89), (d) PKA activator (10 μM, 8-Bromo-cAMP) or vehicle for 30 min at 37℃. AMs were incubated for 1 h with a 3:1 AC:AM ratio or vehicle and challenged with S. pneumoniae (MOI of 50). The lysates were plated onto blood agar for serial dilution quantification of CFU after 24 h. The results are expressed as the percentage of survival of ingested S. pneumoniae compared with control containing S. pneumoniae + AMs (% of control). *P < 0.05 vs. AMs + Sp, #P < 0.05 vs. AMs + AC + Sp.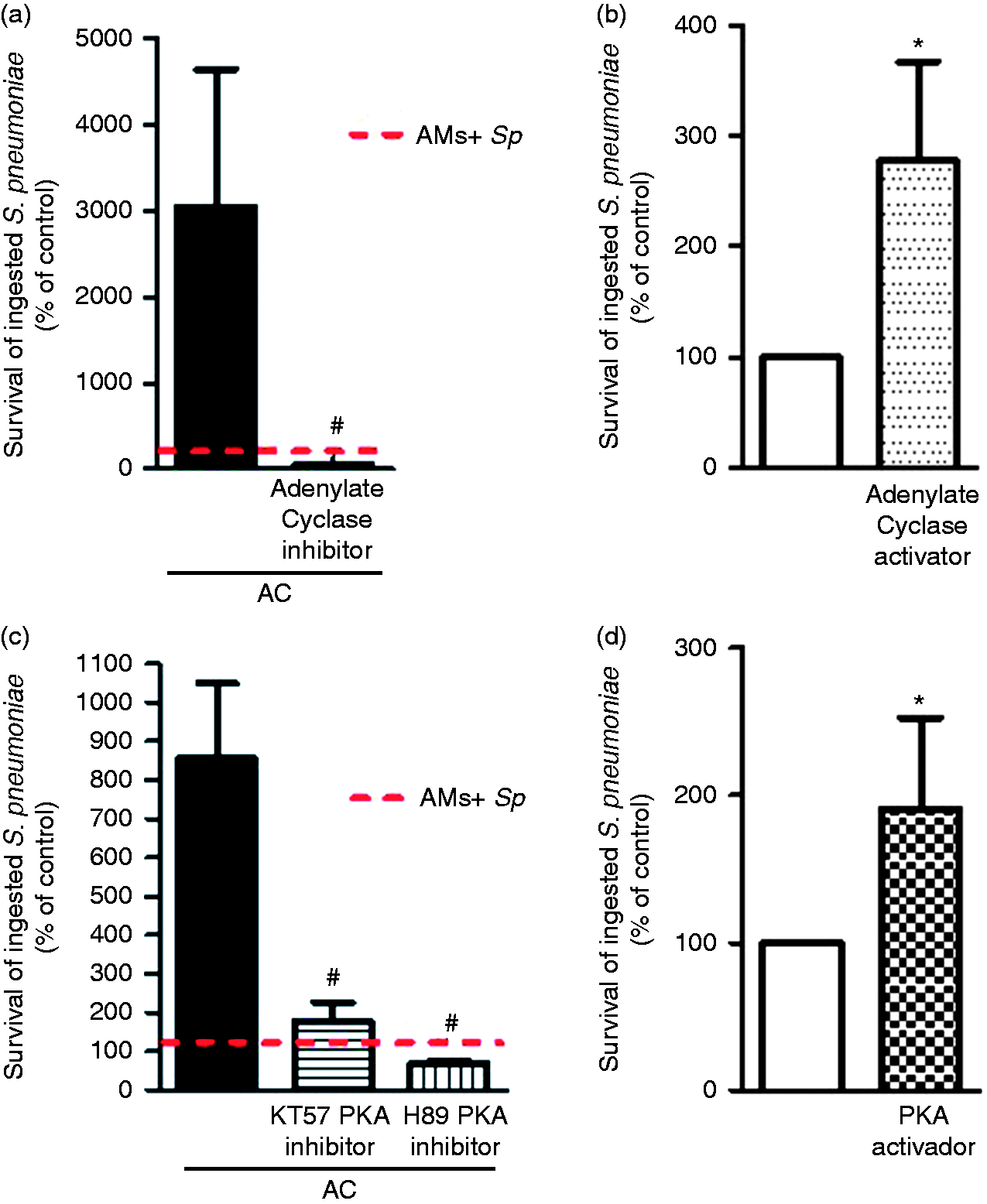
Next, we evaluated whether PKA activation via the increase in cAMP by PGE2/EP2-EP4 accounted for the decreased microbicidal activity detected during S. pneumoniae infection. In the presence of PKA inhibitors KT5720 and H89 dihydrochloride, AMs were able to reverse the inhibitory effects of PGE2 produced during the phagocytosis of apoptotic cells (Figure 3c). To confirm that PKA activation inhibited AM microbicidal activity, the cells were incubated with a PKA activator (8-bromo-cAMP) and challenged with S. pneumoniae, and we found that PKA activation increased bacterial survival within AMs when compared with AMs + Sp (Figure 3d).
Efferocytosis decreases H2O2 production by S. pneumoniae-infected AMs
Next we examined how efferocytosis impaired the killing of S. pneumoniae by assessing whether efferocytosis impairs microbicidal activity by decreasing the levels of NO and H2O2. AMs were co-cultured with or without apoptotic cells and challenged with S. pneumoniae for different periods of time; NO and H2O2 were measured in the supernatant. Low NO levels were detected when AMs were infected with S. pneumoniae. Moreover, there was no difference in NO production by AMs incubated in the presence or absence of apoptotic cells after S. pneumoniae challenge (data not shown). Conversely, H2O2 production was increased after 30 min and 90 min when AMs were infected with S. pneumoniae and the ingestion of apoptotic cells inhibited H2O2 production after S. pneumoniae infection. Furthermore, pre-treatment with different PKA inhibitors was able to revert partially H2O2 production after 30 min; however, this effect was more pronounced after 90 min of incubation of AMs co-cultured with apoptotic cells and challenge with S. pneumoniae (Figure 4). The concentration of PKA inhibitors in H2O2 assay was the same used in microbicidal activity.
Efferocytosis decreases H2O2 production by AMs. AMs harvested from BAL were pre-incubated in the presence or absence of PKA inhibitors (KT5720: 1 μM; H89: 10 μM; PKI6-22: 5 μM) for 30 min, incubated for 1 h with a 3:1 AC:AM ratio or vehicle and challenged with S. pneumoniae (MOI of 50) for 30 or 90 min with 123-dihydrorhodamine (25 μM); the fluorescence intensity was measured. The data are expressed as the percent relative changes in H2O2 production (% AMs + Sp). Representative results of two independent experiments performed in quintuple are shown. *P < 0.05 vs. AMs + Sp, #P < 0.05 vs. AMs + Sp + AC.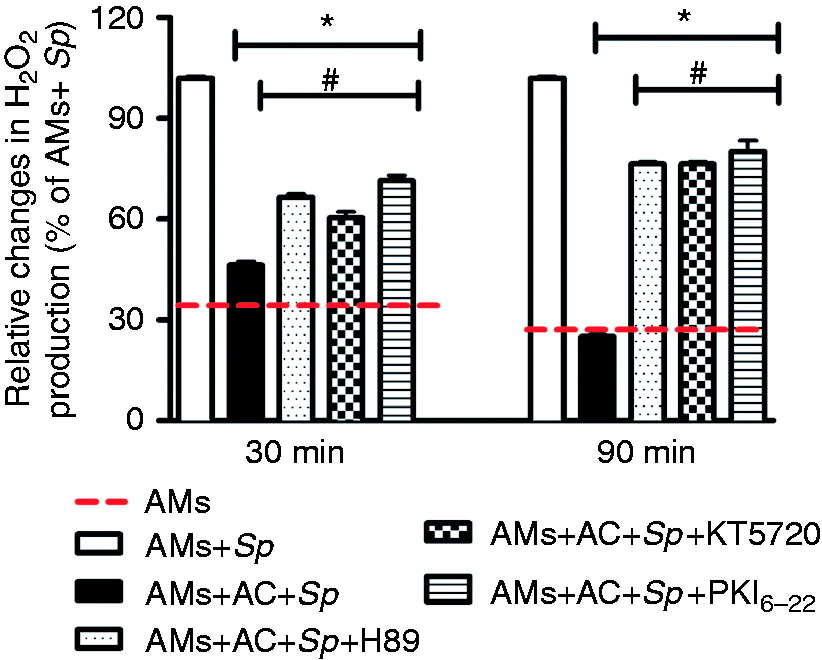
Discussion
In this study, we evaluated whether PGE2 produced during efferocytosis impairs the effector functions of AMs against non-opsonized S. pneumoniae. We found that efferocytosis by AMs had modest inhibitory effects on phagocytosis of S. pneumoniae but greatly inhibited bacterial killing by AMs in a manner dependent on PGE2 production. The deficiency of bacterial killing ability was mediated via the EP2–EP4/cAMP/PKA pathway and inhibition of H2O2production.
Lower respiratory tract infections are among the major causes of death, 20 especially infections caused by Gram-positive bacteria such as S. pneumoniae.21–23 AMs are the most important cells involved in the clearance of apoptotic cells and defense against different microorganisms in the lung. COPD is characterized by defective efferocytosis leading to an intense accumulation of apoptotic cells in the lung and bronchoalveolar space. Moreover, patients with COPD are more prone to developing pneumonia, and S. pneumoniae is the most frequent bacterial lung infection in these patients. 24 We previously demonstrated that efferocytosis by AMs promotes inhibition of FcR-mediated phagocytosis and killing. PGE2 is the major soluble mediator responsible for the deleterious effects observed during efferocytosis. 3 Our results demonstrate that efferocytosis by AMs has a modest inhibitory effect on the phagocytosis of S. pneumoniae. Treatment with the COX inhibitor indomethacin and an adenylyl cyclase inhibitor was able to reverse the inhibition of S. pneumoniae phagocytosis caused by efferocytosis (Figure 1). AMs incubated in the presence of the same ratio of apoptotic cells (3:1) were not able to inhibit the phagocytosis of non-opsonized S. pneumoniae (∼32%), as was previously observed with the Fc receptor (∼60%). Thus, these results suggest that the inhibitory effect of efferocytosis mediated by PGE2 on AMs is less effective on non-opsonic phagocytosis compared with the Fc receptor. 3 IgG–FcR activation induces phosphorylation of ITAM-associated proteins, such as Syk/PI3K and Akt, 25 whereas other receptors such as SR-A lead to activation of the Src family and PI3 kinases AKT/MAPK and Rac. 26 Therefore, efferocytosis by AMs can promote inhibition of phagocytosis by the Fc receptor but has only a weak effect through non-opsonized S. pneumoniae.
Next, we evaluated whether efferocytosis by AMs could affect their microbicidal activity against S. pneumoniae. Ingestion of apoptotic cells impaired bacterial killing, and treatment of AMs with a COX inhibitor or EP2 and EP4 antagonists reversed the inhibitory effects mediated by efferocytosis. Exposure of AMs to PGE2 or an EP2 or EP4 agonist impaired S. pneumoniae killing, with a synergistic effect when AMs were incubated with EP2 plus EP4 agonists. Previously published data support our findings: Aronoff et al. demostrated that EP2 deficiency increased the survival of mice and improved the clearance of S. pneumoniae. 27
We also reported a decrease in bacterial burden in the lung and bloodstream in EP2–/– mice infected with S. pneumoniae compared with WT mice. 3 In the present study, we demonstrate that EP4, in addition to EP2, is involved in the inhibitory effects of PGE2 in AMs. Aronoff et al. 27 suggested that infection with S. pneumoniae induces PGE2 production from cells other than AMs, such as epithelial cells. Interestingly, a recent report demonstrated that S. pneumoniae is able to secrete H2O2, thereby causing DNA damage and inducing apoptosis in epithelial cells and AMs in a manner that is dependent on H2O2 generation. 29 Pneumococcal pneumolysin also promotes apoptosis in the lung epithelium and lung damage during S. pneumoniae infection.30,31 We speculate that S. pneumoniae induces apoptosis, which, in turn, elicits AM efferocytosis, leading to PGE2 production that inhibits bacterial killing. We determined bacterial killing by counting CFUs recovered after infection—we counted both internalized and non-internalized bacteria. While this assay is subject to experimental variations and misinterpretation of the results, every experimental group was treated in the same way and the differences observed were highly reproducible among the different experiments. Furthermore, it is important to note that during the microbicidal assay there was no gentamicin during the wash steps. Recent study has shown that pneumolysin, released by S. pneumoniae, can promote the permeabilization of cell membranes, which could facilitate the access of antibiotics inside the cell and kill intracellular bacteria. 28
PGE2 is able to inhibit effector functions of macrophages and neutrophils, such as H2O2 production. 32 Because the microbicidal function of AMs was inhibited by PGE2–PKA pathway during efferocytosis, we evaluated whether treatment with a PKA inhibitor could increase H2O2 production by AMs during efferocytosis; however, the microbicidal effect of ROS against of S. pneumoniae remains unclear. Here, we demonstrate that AMs challenged with S. pneumoniae produce large amounts of H2O2 and that efferocytosis inhibits production of this microbicidal factor. Nonetheless, exposure to a PKA inhibitor partially reverted the production of H2O2 by AMs during efferocytosis. This result suggests that PKA suppresses the effector function of AMs against S. pneumoniae by a mechanism other than H2O2 production.
Our data show that efferocytosis by AMs inhibits H2O2 production and this inhibitory effect was mediated by PGE2 via the EP2–EP4/cAMP/PKA pathway. To improve the clearance of apoptotic cells by AMs and prevent lung injury, statins or glucocorticoids are a treatment option for COPD, though these patients might become more susceptible to bacterial infection. Therefore, treatment of these patients with COX inhibitors combined with antibiotic therapy may lead to improvement in the immunosuppression caused by PGE2 in the lung microenvironment and restoration of the effector functions of AMs during bacterial infections.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP - 2011/17611-7, 2011/23979-7), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq 471945/2012-9; 306363/2013-5), CAPES, Programa de Apoio ao Desenvolvimento Científico da Faculdade de Ciências Farmacêuticas da UNESP (PADC). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.