
Abstract
We aimed to study the relationship between pain perception and cytokine release during systemic inflammation. We present a randomized crossover trial in healthy volunteers (n = 17) in 37 individual trials. Systemic inflammation was induced by an i.v. bolus of Escherichia coli LPS (2 ng/kg) on two separate trial days, with or without a nicotine patch applied 10 h previously. Pain perception at baseline, and 2 and 6 h after LPS was assessed by pressure algometry and tonic heat stimulation at an increasing temperature (45–48℃) during both trials. Compared with baseline, pain pressure threshold was reduced 2 and 6 h after LPS, while heat pain perception was accentuated at all testing temperatures after 2 but not 6 h. The magnitude of changes in pain perception did not correlate to cytokine release. No effect of transdermal nicotine or training status was observed. In conclusion, LPS administration in healthy human volunteers leads to reduction in pain pressure threshold and an increase in pain perception to heat stimuli, supporting a relationship between acute systemic inflammation and pain perception.
Keywords
Introduction
Systemic inflammation is a generalized response to trauma, surgery or severe infection, 1 and almost ever-present in the critically ill. 2 The human endotoxin model has been employed for more than three decades in order to study, in particular, the early phase of the systemic inflammatory response. 3 In this model, an i.v. bolus injection or infusion of Escherichia coli LPS (also referred to as endotoxin), a purified, non-infectious component of the outer cell envelope of most Gram-negative bacteria, is administered to healthy volunteers. A dose-dependent, fully reversible response is evoked with a duration of a few hours, during which flu-like symptoms and increased levels of pro- and anti-inflammatory markers are observed. 4 The model is not an accurate replication of acute infection, the systemic inflammatory response syndrome or critical illness in clinical practice; however, it induces a reproducible, inter-individually comparable and fully reversible inflammatory response, which allows for experimental studies in humans of early stages of acute systemic inflammation and in the interplay between different aspects of critical illness complicated by systemic inflammation. 4
An inflammatory-induced state of hyperalgesia, i.e. increased pain perception, in this context is well documented in both animal and human experimental studies, including those using the human endotoxin model.5–14 Most likely, hyperalgesia is also present during systemic inflammation in the critically ill and may contribute to the well-recognized problem of pain in these patients. Whereas the pathogenesis of hyperalgesia during systemic inflammation is poorly understood, the close relationship between inflammation and pain perception suggests a dose–response relationship, where a greater inflammatory response leads to an enhanced pain perception, and existing evidence supports this notion. 11 Though heterogenous in design, previous studies support the notion of intensified pain perception during inflammation,7–14 but the level of inflammation needed to induce hyperalgesia has not been established. Also, the time course of the altered pain perception has not been clearly defined, with the onset and peak being more intensively studied than the remission. No previous studies have evaluated the reproducibility of inflammatory-induced hyperalgesia through repeated LPS exposure.
The present study was part of a trial evaluating the effect of training status and nicotine administration on systemic inflammation in the human endotoxin model. As pro-inflammatory cytokine release from macrophages is inhibited by vagus nerve activity via the nicotinergic α7-subtype acetylcholine-receptor α7nAChR, 15 the primary aim of the core trial was to study the inflammatory response in well-trained compared with untrained healthy volunteers with perceived high and low vagal activity, as well as the effect of transdermally administered nicotine on this response. The design of two study days provided us with an opportunity to study pain perception during repeated LPS exposure and during an extended time compared with previous studies. Thus, we hypothesized that pain perception in healthy individuals would be increased after LPS, and that the magnitude of the inflammatory response, as measured by plasma cytokine levels, would correlate with changes in the aforementioned indices of pain perception. However, we did not intend to address the effect of training status on pain perception.
Materials and methods
This study was part of a project evaluating the effect of training status and nicotine administration on systemic inflammation in the human endotoxin model as described above. The study protocol was approved by the Regional Committee on Health Research Ethics (protocol-ID H-3-2012-011) and the Regional Data Monitoring board (ID j-2007-58-0015, local 30-0766) and registered on clinicaltrials.gov (NCT01592526). The study was conducted in accordance with the principles of the Declaration of Helsinki, and oral and written informed consent was obtained from all participants.
Neither these data nor data from other parts of this study have been published or submitted for publication previously.
Participants
Healthy, non-obese, non-smoking males aged 18–35 yr were recruited into two groups: well-trained (VO2max ≥ 60 ml O2/kg/min) or untrained (VO2max ≤ 45 ml O2/kg/min). These cut-off values were based on the normative data published by Cooper 16 and confirmed in a study of fit and sedentary young men. 17
Pre-study screening included self-reported medical history, physical examination, routine laboratory tests, ECG, dual X-ray scanning and an ergo-metric bike-test (Monark, Vansbro, Sweden) for measurement of maximal oxygen consumption (VO2max).
Design
This was an open-label, randomized crossover study with two separate study days (trial A and B, respectively), on which an identical dose of LPS was injected. In order to address the research questions of the core trial, trial B was preceded by the application of a nicotine patch (15 mg/16 h, Nicorette® invisi; Johnson & Johnson, New Brunswick, Canada), applied 10 h before and removed 6 h after the LPS challenge. The sequence of trials A and B was randomized. To minimize the risk of tolerance, a minimum interval of 4 wk was applied between study days. Furthermore, to avoid any effects of recent infection, experiments were cancelled if the patient had experienced fever or malaise within the last 2 weeks before the trial days. Finally, the patients were instructed to remain physically inactive for 24 h prior to each trial day.
On both trial days, participants reported to the laboratory following an overnight fast. Two peripheral i.v. catheters were inserted for injection of LPS and blood sampling. Following a 2-h baseline period, a bolus injection of LPS (2 ng/kg, Lot EC-6; National Institutes of Health, Bethesda, MD, USA) was administered i.v. Vital signs (non-invasive blood pressure, heart rate, pulse oximetry and temperature) were recorded and blood samples for measurement of plasma TNF-α, IL-6, IL-8, IL-10, IL-1β and cortisol were collected at −2, −1, 0, 1, 1½, 2, 3, 4, 5 and 6 h, where t = 0 denotes the time of LPS injection. Furthermore, pain pressure threshold and pain intensity with heat stimulation as described below were assessed as described below at −2, 2 and 6 h in all individual 37 trials (Figure 1).
Flowchart: assessment, inclusion and withdrawal of participants.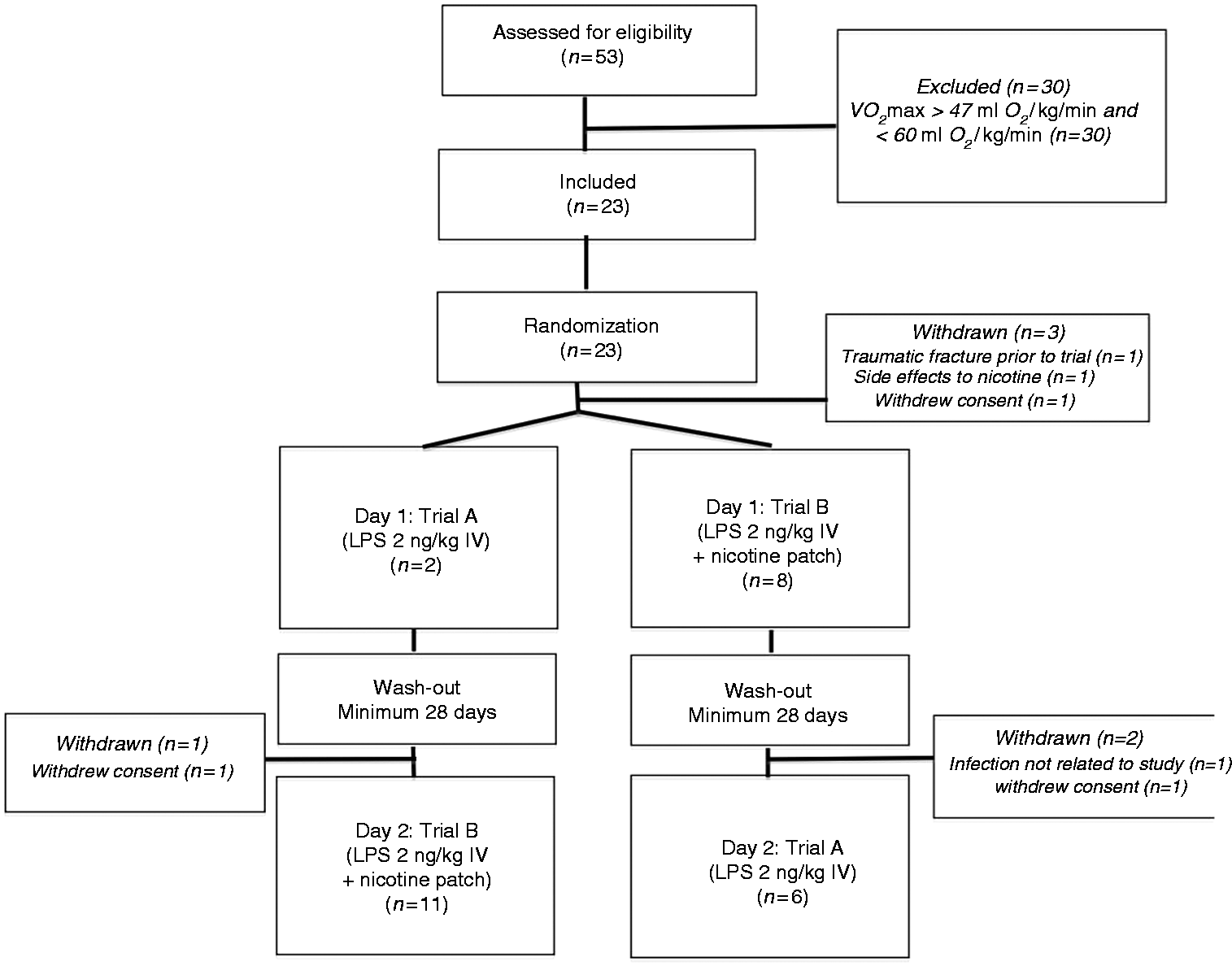
Randomization procedure
Sequence allocation was block-randomized by the participants’ fitness level (well-trained or untrained). For each of these two groups, a box containing 12 notes in opaque envelopes was produced. Six notes in each box assigned the participant to the sequence ‘nicotine before control’ and vice versa. A staff member, who did not otherwise participate in the study, drew an envelope from the relevant box after informed consent had been obtained. The assignment was unblinded.
Pressure algometry
Pressure algometry was performed by a hand-held device (Somedic AB, Hörby, Sweden) with a tip area of 1 cm2 applied perpendicularly on the non-dominant lateral vastus muscle. Pressure was gradually increased (30–40 kPa/s) and the participant was told to indicate by a handheld button when the pressure was perceived as painful. The pressure pain threshold (PPT) was assessed in triplicate and the mean value was used in the data analysis. All tests were conducted by one person (SJ) and participants were blinded to the test results.
Heat stimulation
Heat stimulation was performed using a computerized contact thermode (active area 2.5 × 5.0 cm2; Thermotest, Somedic AB, Hörby, Sweden), which was applied on the mid-sagittal line of the non-dominant thigh, 20 cm above the upper edge of the patella with the knee held in a 90° flexion. The baseline (thermoneutral) temperature was 32℃ and a ramp rate of ± 2℃/s was used. A sequence of four tonic heat stimuli (45, 46, 47 and 48℃) was delivered with a duration of 5 s each and separated by an interstimulus interval of 30 s at baseline temperature. Participants rated each heat stimulus immediately after stimulation with a slider on a visual analogue scale (VAS) anchored by 0 and 10 (VAS; 0 = no pain, 10 = worst imaginable pain).
Pressure algometry and supra-threshold heat stimulation have been used in a number of human experimental pain studies using endotoxin-induced inflammation.9,11–13
Measurement of plasma cytokines and cortisol
Blood samples for analysis of cytokine levels in plasma were collected in EDTA tubes (Greiner bio-one, Germany). The samples were kept on ice until centrifuged at 4℃ and 1233 g (at Rmin) for 10 min, and the supernatant was stored at −80℃ until analysis. Concentrations of TNF-α, IL-6, IL-8, IL-10 and IL 1β were analyzed using ELISA (Meso Scale Discovery, Rockville, Maryland, USA).
Blood samples for analysis of plasma cortisol level were collected in lithium heparin containing tubes (Greiner Bio-One, Frickenhausen, Germany) and analyzed using a sandwich electro-chemiluminescence immunoassay (Roche, Basel Switzerland) at the Department of Clinical Biochemistry, Rigshospitalet, Denmark.
Statistical analysis
The sample size calculation for this pilot study was conducted according to the primary objective of the core trial, which was to measure the inflammatory response according to training status. This calculation was based on estimates of the area under the curve (AUC) for plasma TNF-α during the control situation and after transdermal administration of nicotine. In a paired t-test, n = 3 would allow us to demonstrate an effect, provided all participants experienced the same effect. When comparing trained and untrained individuals, n = 12 would detect a difference of 1.2 × SD between the two groups with a power of 80% and α = 0.05. Prior studies found a mean ± SD peak TNF-α level in plasma of 1260 ± 660) pg/ml. 18 Under this assumption, we would be able to detect a difference in plasma TNF-α of 800 pg/ml or approximately 60% of the expected mean value.
Data were tested for normality using the Shapiro–Wilks test.
Because neither nicotine administration nor training status affected the inflammatory response or pain perception in subjects, as described in the ‘Results’ below, data were pooled and reported regardless of nicotine or training status.
Individual data on single trial days (time-course) were assessed using one-way ANOVA followed by paired t-tests to identify points of significance.
Changes in pain pressure threshold, VAS scores for heat perception and plasma cytokine release (expressed as the AUC) within subjects on alternate trial days were tested using Student’s paired t-test. Differences in values between well-trained and untrained subjects were analysed using an unpaired t-test. Correlation analysis was performed using Pearson’s rank R correlation. Data are expressed as mean (95% confidence interval [CI]) unless otherwise stated.
Statistical calculations and graphs were performed using GraphPad Prism V5.0 (GraphPad Software, La Jolla, CA, USA). Differences were considered statistically significant at P < 0.05.
Results
Twenty-three volunteers were included in the study. Of these, 20 participants completed one trial day, and 17 of these completed both study days (flowchart, Figure 1). For the latter group, the median interval between d 1 and d 2 was 42 d (range 28–77 d). As depicted in Figure 1 three volunteers withdrew from the study after being formally included and randomized, but before d 1. Two volunteers withdrew their consent to participate; one stated side effects to the nicotine patch as a reason and withdrew prior to the first study day, and one reported no reason. Also, one volunteer was excluded owing to a traumatic fracture. These three volunteers yielded no data for the study and therefore never entered data analysis.
Characteristics of participating groups (well-trained vs. untrained).
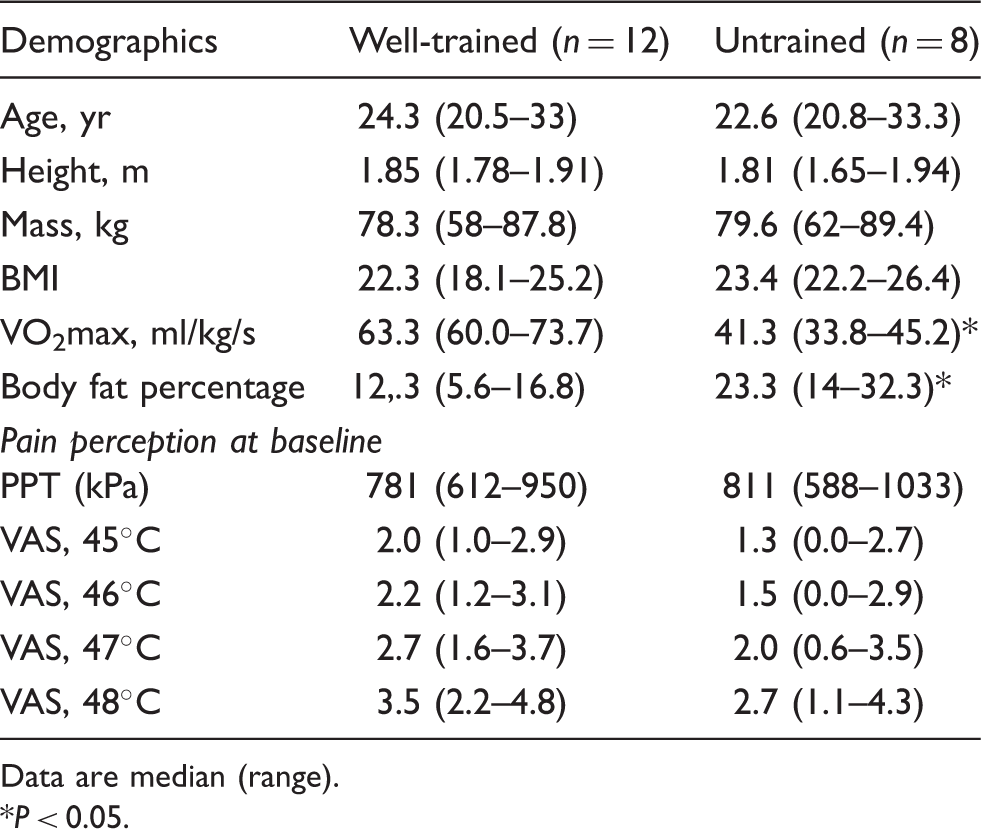
Data are median (range).
P < 0.05.
As no effect of training status or nicotine administration was observed with respect to the cytokine and cortisol response and to pain perception after LPS (all P-values > 0.10), data were pooled and results are reported for all volunteers, irrespective of training status.
Inflammatory response to LPS
On both trial days, injection of LPS (2 ng/kg) induced flu-like symptoms (malaise, shivering, muscle pain, nausea, headache and dizziness), as well as an increase in body temperature. Cytokines (TNF-α and IL-6) increased at 60 min and peaked at 90 and 120 min, respectively, after LPS (Figure 2). Symptoms resolved and cytokines normalized within 6 h of administration.
TNF-α and IL-6 after bolus LPS (2 ng/kg) i.v. Total cytokine release expressed by the AUC was reduced on the chronologically second trial day for both TNF-α (P < 0.01) and IL-6 (P < 0.001).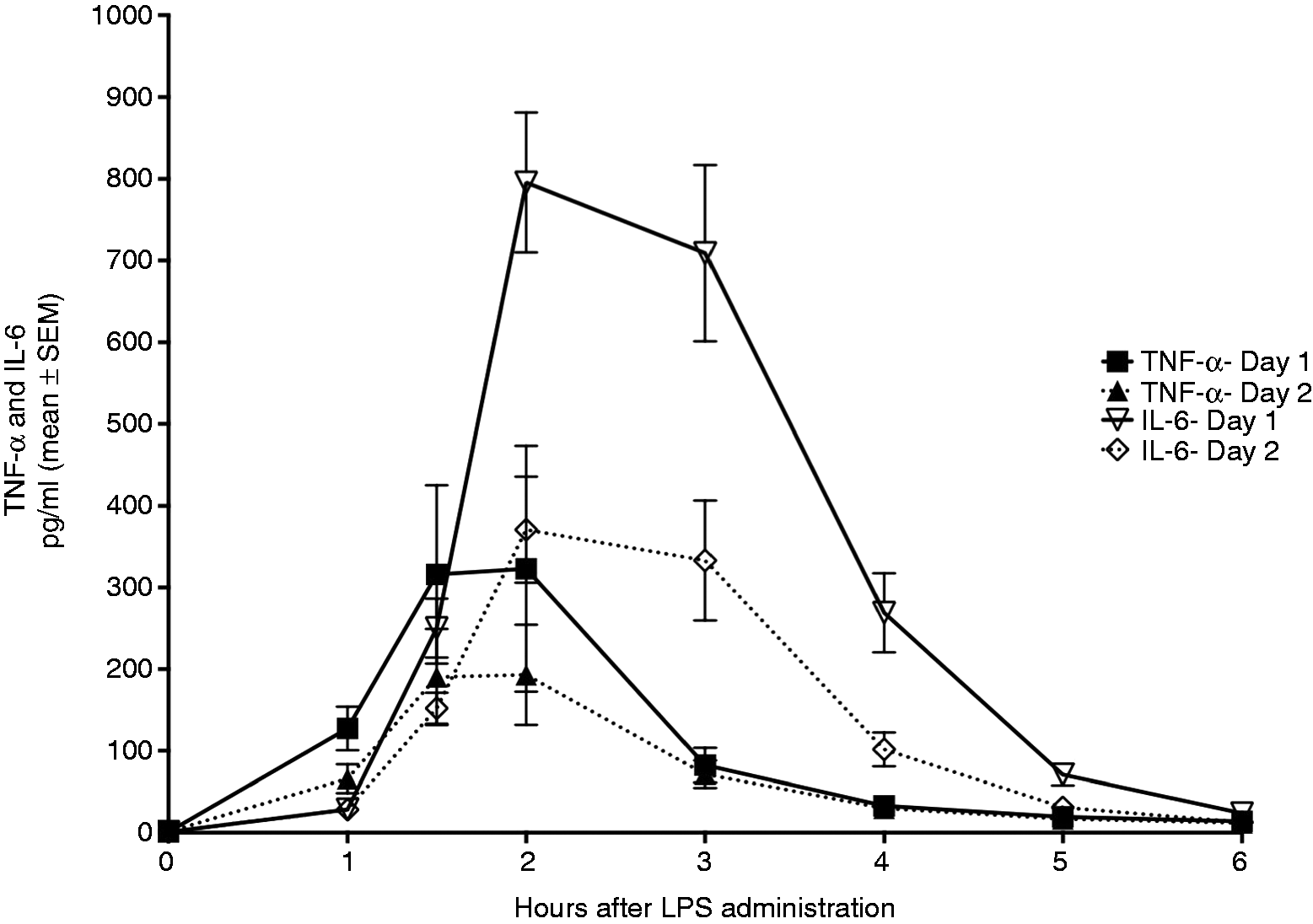
Cytokine release was reduced on d 2 compared with d 1, consistent with the development of tolerance, regardless of allocation sequence (nicotine or control on the first trial day) or training status (Figure 2). Thus, peak TNF-α level declined from 323 ± 139 pg/ml on d 1 to 193 ± 57 pg/ml on d 2 (P = 0.10), while peak IL-6 declined from 796 ± 168 pg/ml to 371 ± 127 pg/ml (P = 0.002), respectively. Also, total release of TNF-α, expressed by the AUC, declined from 637 ± 187 to 417 ± 127 pg/ml h, i.e. a reduction of 221 ± 146 pg/ml h or 24 ± 16% (paired t-test, P < 0.01), respectively. Similarly, total release of IL-6 was reduced from 2109 ± 517 pg to 1347 ± 366 pg/ml/ h, equal to a reduction of 761 ± 362 pg/ml h or 30 ± 14% (paired t-test, P < 0.001) (Figure 2).
Cortisol increased at 60 min and peaked 4 h after LPS administration. Neither training status (P = 0.86) nor nicotine administration (P = 0.87) had any effect on cortisol levels. In accordance with the observations above, there was a trend towards a reduced cortisol release on d 2. Peak levels declined from 953 ± 56 nmol/l to 873 ± 66 nmol/l (P < 0.05) and total release, expressed by the AUC, decreased from 5008 ± 291 nmol/ml/h on d 1 to 4637 ± 210 nmol/ml/h (P = 0.06) on d 2.
Quantitative sensory testing
In the following, results from measurements on chronological d 1 (n = 20) will be described, if not clearly stated otherwise.
Pressure algometry
After LPS, the PPT was reduced both at 2 and 6 h (change from baseline, −151 kPa (−221 to −82 kPa) and −149 kPa (−218 to −78 kPa), respectively (P < 0.0001 for both, compared with baseline) (Figure 3a).
(a) PPT after bolus LPS (2 ng/kg) i. v. *P < 0.0001, compared with baseline (one-way ANOVA, Bonferroni multiple comparisons test). (b) PPT after bolus LPS (2 ng/kg) i.v. A slight increase in pain pressure threshold was observed at baseline on trial d 2 (P < 0.01, Student’s paired t-test). Neither absolute nor relative change in threshold from baseline to 2 h and 6 h differed from that observed on trial d 1.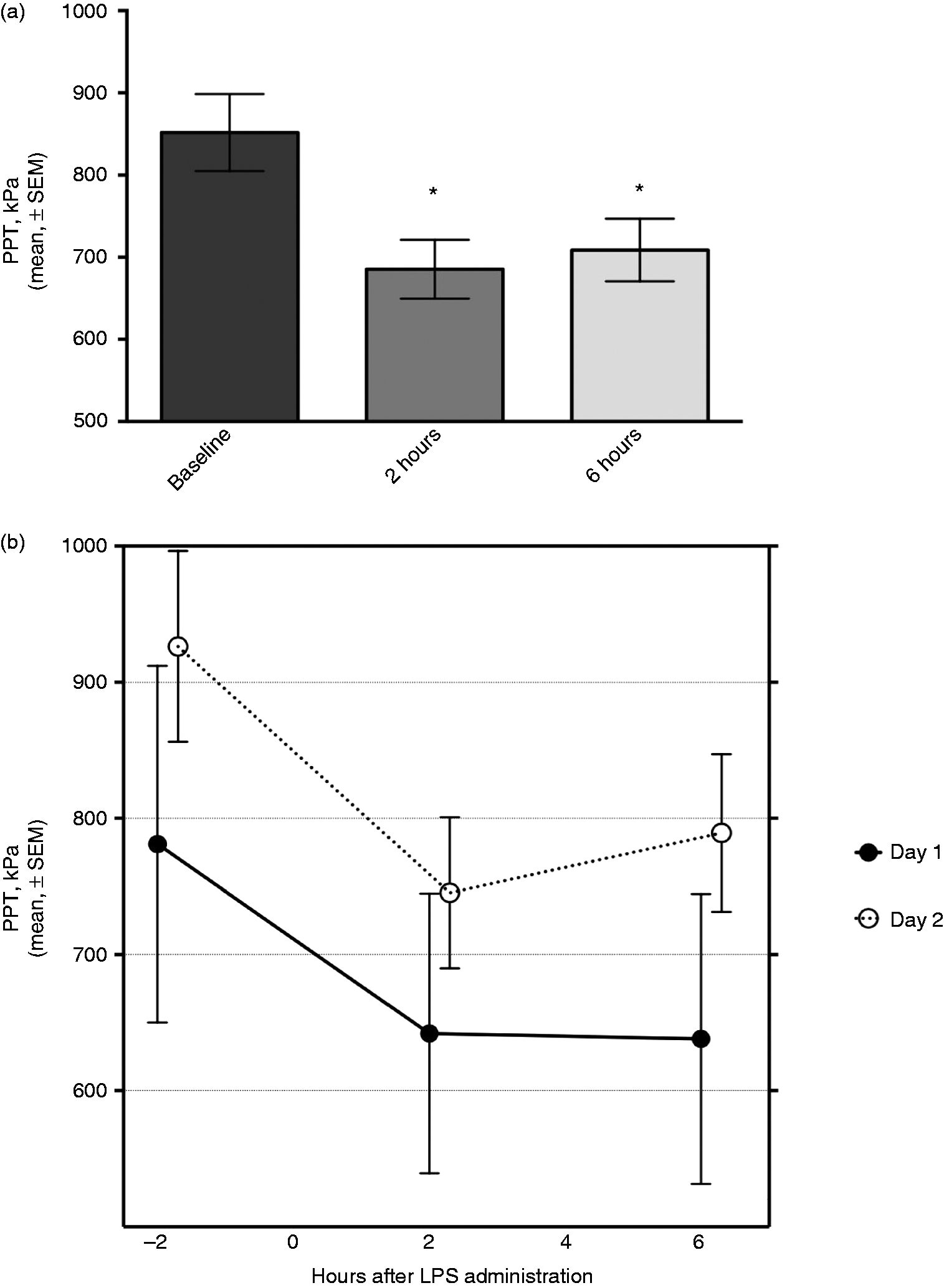
Heat stimuli
Compared with baseline values, heat pain perception was enhanced at all temperatures at 2 h [VAS change, 2 h vs. baseline at 45℃: 0.7 (0.0–1.4); 46℃: 1.1 (0.4–1.8); 47℃: 1.3 (0.6–2.0); and 48℃: 1.4 (0.8–2.1)] but not at 6 h after LPS administration (Figure 4).
(a) Pain intensity after bolus LPS (2 ng/kg) i.v. Pain perception in response to standardized heat stimuli was increased 2 h after bolus LPS injection as measured by VAS (45℃, P < 0.01; 46℃, P < 0.01; 47℃, P < 0.001; 48℃, P < 0.001, repeated measures ANOVA, Bonferroni multiple comparisons test). Six h after bolus LPS injection no significant change from baseline was observed. (b) Pain intensity after repeated exposure to bolus LPS (2 ng/kg) i.v. Heat pain perception was highly reproducible at baseline, while the increase at 2 h was slightly attenuated on d 2 at temperatures 47℃ (P < 0.05) and 48℃ (P < 0.05).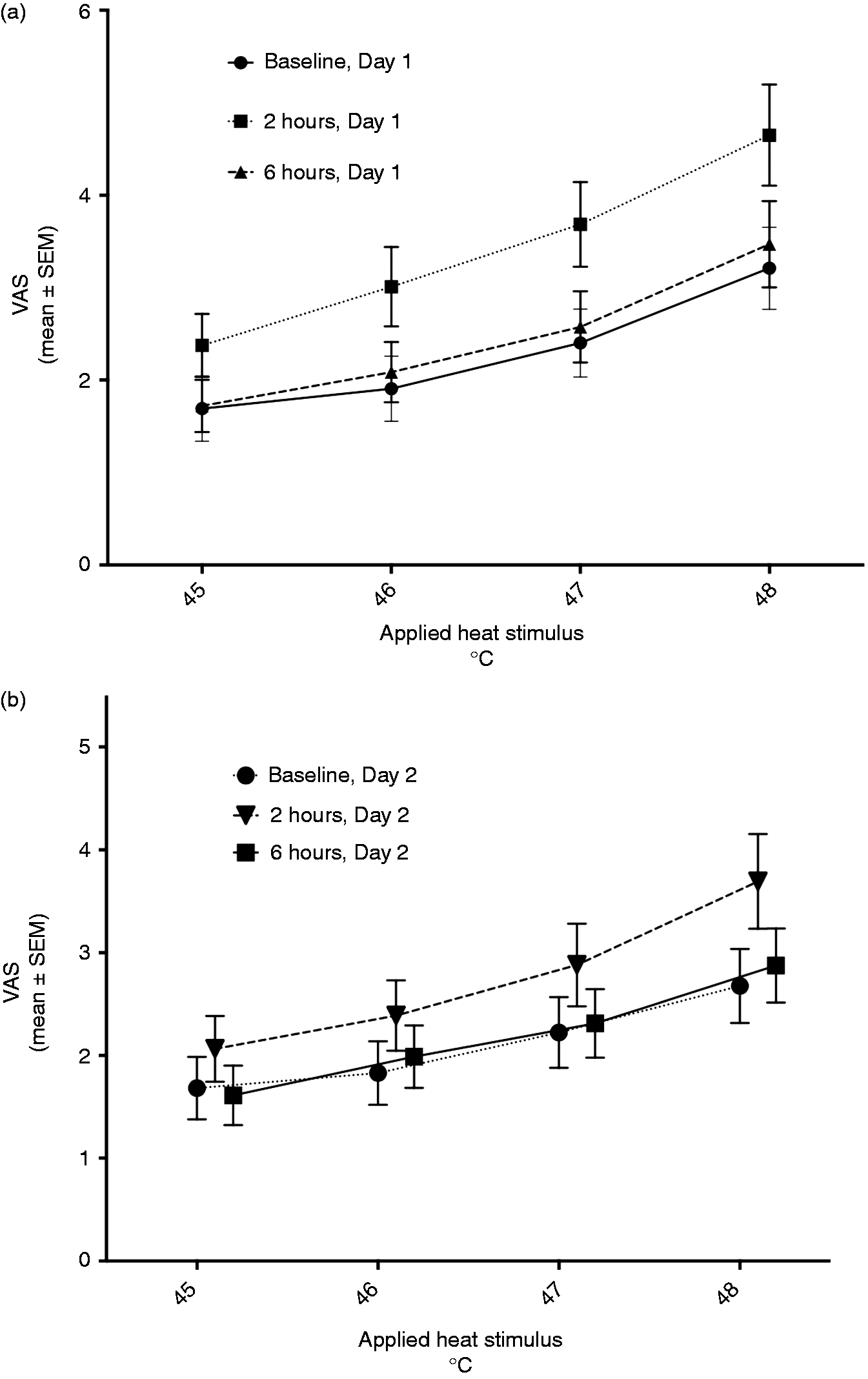
Pearson’s correlation coefficients between peak levels of plasma cytokines and changes from baseline in pain pressure thresholds and heat pain perception 2 h after injection of E. coli LPS in healthy young men.
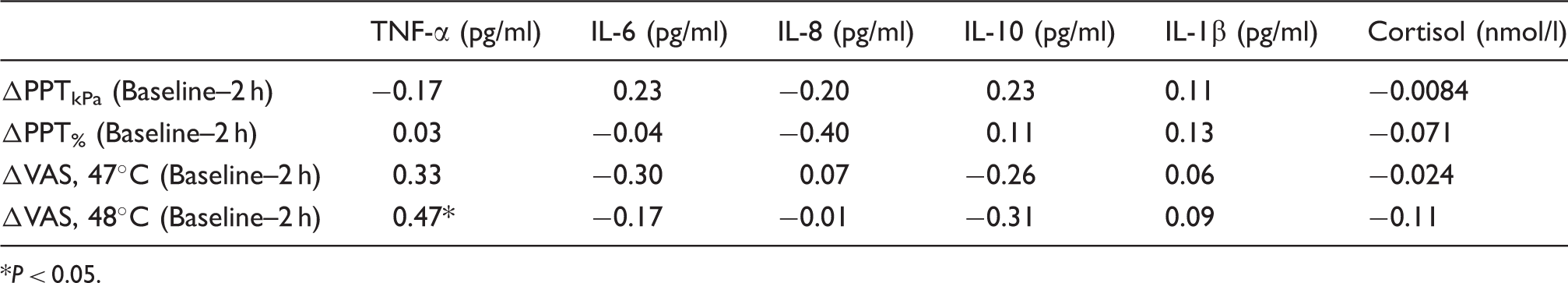
P < 0.05.
Repeated LPS exposure
On d 2 compared with d 1, a slight increase in baseline PPT was observed [mean difference, 144 kPa (47–242 kPa), paired t-test, P < 0.01]. Neither absolute nor relative changes in threshold from baseline to 2 h and 6 h differed from those observed on d 1 (Figure 3b).
Heat pain perception was highly reproducible at baseline (mean differences ranging from −0.49 to 0.018 VAS units; all P-values > 0.10) and at 6 h compared with d 1 (mean differences ranging from −0.70 to −0.04 VAS units; all P-values > 0.10), while the increase at 2 h was slightly attenuated on d 2 at temperatures 47℃ [mean difference, −0.74 (−1.47 to −0.001) VAS units; P < 0.05] and 48℃ [mean difference, −0.91 (−1.7 to −0.1), P < 0.05] (Figure 4b).
Discussion
In the present study, we found a change in pain perception during experimental systemic inflammation in humans. Thus, the PPT was reduced and the VAS score to cutaneous heat stimulation was increased at 2 h after LPS. We observed a rapid recovery towards baseline values at 6 h for heat perception but not for the PPT. The change in pain perception appeared not to be directly related to the measured concentrations of circulating cytokines, as indicated by the correlation calculations. Neither training status nor nicotine treatment appeared to affect cytokine release or pain perception, but the study has insufficient power to draw any firm conclusion regarding this issue. Contrary to our expectations, we observed evidence of tolerance with attenuation of the inflammatory response after repeated LPS administration with a median interval of 42 d (range 28–77 d) after the first bolus.
The human endotoxin model mimics the early phase of acute systemic inflammation with good inter-individual reproducibility, and it is a safe procedure for the volunteers, given its absence of tissue injury, and it has full reversibility. Thus, at least in theory it is suitable to address the potential relationship between the inflammatory response and pain perception. Our findings using this model agree with those reported by de Goeij et al., 12 who used an identical LPS dose and measured PPT, electrical pain threshold and the cold pressor test. This study found enhanced pain perception at 2 h after LPS, and also a lack of correlation between the change in pain perception and cytokine release. Other studies of pain and inflammation have induced ‘low-grade inflammation’ using LPS doses of 0.4–0.8 ng/kg. In this setting, Wegner et al. 11 attempted to establish a dose–response relationship through quantitative sensory threshold measurements in three groups of volunteers, who received LPS 0.8 ng/kg, LPS 0.4 ng/kg or saline. 11 In that study, reduced PPTs were found at all anatomical sites (lower back, calf, suprapinatus muscle and deltoid region) with the higher dose of LPS, but only over the lumbar region with the lower dose (0.4 ng/kg). In contrast, mechanical pain sensitivity and cold pressor test were unaffected by LPS. Also, Hutchinson et al. 8 reported that LPS administration enhanced hyperalgesia after intradermal capsaicin injection. The effect of low-grade inflammation on visceral pain was addressed by Benson et al., 10 who reported a reduced pain threshold to rectal distension during peak inflammation. Finally, although Karshikoff et al. 9 found a reduction in pain threshold to heat and cold in women, but not in men, this result was contradicted by Wegner et al., 13 who did not observe any sex-related differences in somatic or visceral pain perception. Another study, also by Wegner et al., 13 designed to address sex-related differences found no evidence of differences in inflammatory-induced pain sensitization between men and women, although the group of female volunteers revealed a more pronounced pro-inflammatory response with peak levels of cytokines (TNF-α and IL-6) and cortisol significantly higher than those of their male counterparts in response to LPS 0.4 ng/kg.
Contrary to our hypothesis, the cytokine response, as measured by the AUC of plasma levels, did not correlate with pain perception. Interestingly, previous studies of experimental low-grade inflammation induced by low-dose LPS have reported a modest correlation between the inflammatory response to LPS and pain perception in humans. Thus, Benson et al. 10 reported a correlation between IL-6 and PPT on examination of visceral pain at 1 h. 10 Karshikoff et al. 9 reported a positive correlation between circulating levels of IL-6 and IL-8 and pain intensity in response to heat stimuli 3.5 h after LPS injection, 9 while Wegner et al. found a correlation between IL-6 and PPT reduction in one study, 11 but less so in a different study. 13 In contrast, in the high-dose LPS study by de Goeij et al., 12 no correlation was found.
The present study extends these previous findings by reporting that different aspects of pain perception follow different rates of recovery after a standardized inflammatory stimulus. Moreover, the PPT and heat pain perception were similarly affected after LPS on both trial days, despite the attenuated cytokine release on d 2. Both these observations appear to corroborate the notion that circulating cytokines do not directly mediate the change in pain perception during systemic inflammation. We speculate that although the inflammatory response is clearly the igniting event that leads to changes in pain perception, circulating cytokines may play a limited role at mediating these changes. Alternatively, the low levels of cytokines induced by low-dose LPS are, in fact, sufficient to mediate an enhanced pain perception, but the presence of a ceiling effect indicates that higher cytokine levels do not contribute further to this effect as seen in this study and the study by de Goeij et al. 12
The finding that PPT was higher at baseline on d 2 probably reflects a habituation response: individuals were familiarized with the experiment and the surroundings and generally more comfortable on d 2.
While circulating cytokines may not be involved in pain perception, locally acting cytokines that are governed by locally upregulated mediators may still play a part. Most notably, the role of IL-1β in pain facilitation has been extensively studied (reviewed by Ren and Torres 19 ), suggesting both direct peripheral nociceptive activity through direct action on nociceptive fibres, 20 and indirect peripheral activity through upregulation of pro-nociceptive mediators. 21 The majority of studies in this area are, however, either in vitro or animal experiments studying local mechanisms. 19
The exact mechanism for the initiation or maintenance of hyperalgesia by the inflammatory mediators are not known, but it is probable that several anatomical sites are involved: the peripheral nervous system, the dorsal root ganglia and CNS, for example at the microglial level. 22 As the present model in volunteers does not involve inflammation in the peripheral nervous system, the effect is most probably mediated at the central microglia level. 22
A central mechanism of pain facilitation has been proposed in relation to human endotoxemia. 6 It is closely related to the widely acknowledged theory that generalized hyperalgesia is a central phenomenon,23,24 and is to some extent supported by the finding that negative mood alterations rather than magnitude of cytokine release are correlated to altered pain perception. 11 A recent study by Benson et al. 14 showed enhanced visceral pain-induced neural activation within the dorsolateral prefrontal cortex in an experimental setting where functional magnetic resonance imaging was combined with LPS administration (0.4 ng/kg) and visceral pain threshold determination through rectal distension. CNS effects following endotoxin exposure are well documented regarding neuroendocrine stress response, 25 cerebral blood flow and metabolism18,26 and autonomic nervous activity, 27 yet the exact mechanisms are unclear. Given the size of the LPS molecule it is highly unlikely that it crosses the blood–brain barrier, and earlier studies have demonstrated no net flux of cytokines from blood to brain. 18 An afferent vagal pathway has been proposed 28 but remains poorly understood. The present study was not designed to study central nervous aspects of pain perception, and further elaboration on any such mechanism awaits further studies, but it seems likely that inflammatory-induced hyperalgesia is a complex, multi-facetted phenomenon.
Strengths and limitations
The human endotoxin model is a well validated and standardized model, and all measurements were performed strictly according to a protocol rendering readily comparable data on pain perception during systemic inflammation. All individuals were fasted and subjected to bed rest; all other interventions (fluid infusion, i.v. catheterizations, blood sampling, monitoring) and stimuli (tests of autonomic nervous function, information level, personnel, physical surroundings) were standardized with a minimal risk of bias. The majority of volunteers returned for a second trial day with completely similar pain perception results in the face of an attenuated inflammatory response, clearly demonstrating the reproducibility of data. The unanticipated attenuation of the inflammatory response, however, also represents a weakness in this study. Some level of tolerance to repeated LPS exposure has been reported previously,4,29,30 with shorter interval between studies, but crossover studies using endotoxin have been performed before with no evident carry-over effect when using a 4-wk ‘wash-out’ phase between study days. 31
Another possible limitation in interpretation of these results is the difference in aerobic capacity and body composition between participants. We intended to exploit this variation to test the hypothesis of the core trial that a higher aerobic capacity would be associated with an attenuation of the inflammatory response due to increased vagus-mediated signalling. These characteristics did not appear to affect pain perception neither in this study nor in other comparable studies, but the study was not designed and more importantly not powered to conclude on this subject.
The application of a nicotine patch on alternate occasions in this study represents a possible bias. The effect of nicotine on pain perception has been investigated in numerous studies, but results are inconsistent. 32 One reason may be that most studies investigate regular or occasional smokers, who probably display various degrees of habituation following chronic exposure to nicotine. Altered pain perception before and after nicotine exposure in smokers, may reflect stages of abstinence and satisfaction, rather than direct antinociceptive effects of nicotine. For example, in a population of smokers Pomerleau et al. 33 showed delayed pain awareness and increased pain threshold to cold exposure following nicotine exposure after a period of abstinence. Other studies investigate event-related potentials in habituated smokers after nicotine exposure, with conflicting results.34–37 Of interest, the study by Miyazaki et al. 37 suggested antinociceptive effects of nicotine using event-related potentials, yet no significant changes in subjective pain perception (VAS) were identified. To summarize, there is no evidence demonstrating a direct antinociceptive effect of nicotine in non-smokers with no smoking history. Our paired baseline data do not indicate any such effect and we consider the risk of bias related to this minimal.
Finally, the study was unblinded and there was no placebo control group. Participants knew if they were subjected to nicotine administration or not, and that they received LPS on both trials days and what to expect from this exposure, and they were informed about tests of pain perception and the sequence of heat stimuli applied before the study was conducted. They were also informed about expected results (reduced pain threshold and intensified pain perception) during systemic inflammation; however, they were blinded to their own previous and current scores in both PPT and during heat stimuli.
Conclusion
Following injection of LPS in healthy human volunteers, we observed a reduction in pain pressure threshold and an increase in pain perception to supra-threshold heat stimuli. Neither transdermal nicotine administration nor training status affected pain perception scores. No consistent correlation was found between changes in pain perception and circulating cytokine levels.
These data strongly support a relationship between acute systemic inflammation and pain perception, which appears not to be mediated by the level of circulating cytokines.
Footnotes
Acknowledgments
We are in debt to the Department of Anesthesiology, Bispebjerg Hospital, Copenhagen, who generously supported the study by engaging Susanne Janum, MD for 6 months, while she was preparing and working on this project. We are grateful to Ruth Rovsing, Centre of Inflammation and Metabolism, 7641, Rigshospitalet, Copenhagen and the Department of Clinical Biochemistry, Rigshospitalet for excellent technical assistance.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This article was financially supported by Grosserer Ehrenreichs Foundation (214,000 DKK), Aase and Ejnar Danielsens Foundation (150,000 DKK), The Danish Society of Anesthesiology and Intensive Care Medicine (DASAIM) (75,000 DKK), The Medical Society of Copenhagen (50,000 DKK) and Kong Christian d. 10.’s Foundation (10,000 DKK).