
Abstract
Chronic obstructive pulmonary disease (COPD), a major cause of mortality and morbidity worldwide, is considered an archetypical disease of innate immunity, where inhaled particles and gases trigger an inflammatory response, favoring tissue proliferation in small airways and tissue destruction in lung parenchyma, in addition to the recruitment of immune cells to these compartments. Although cigarette smoking is still considered the main risk factor for developing COPD, the trend of proposing biomass smoke (BS) exposure as a principal risk factor is gaining importance, as around 3 billion people worldwide are exposed to this pollutant daily. A considerable amount of evidence has shown the potential of BS as an enhancer of lung inflammation. However, an impairment of some innate immune responses after BS exposure has also been described. Regarding the mechanisms by which biomass smoke alters the innate immune responses, three main classes of cell surface receptors—the TLRs, the scavenger receptors and the transient receptor potential channels—have shown the ability to transduce signals initiated after BS exposure. This article is an updated and comprehensive review of the immunomodulatory effects described after the interaction of BS components with these receptors.
Keywords
Introduction
Chronic obstructive pulmonary disease (COPD) is a highly prevalent condition globally, with important repercussions on the rate of mortality and high costs of care. Global estimates place COPD as the fourth leading cause of death worldwide and it is expected to become the third in 2030, 1 a position that it has already reached in the USA. 2 COPD is characterized by an abnormal response of the lungs to toxic particles and gases, resulting in a progressive and not fully reversible airway obstruction whose main pathologic hallmarks are bronchiolitis and lung parenchymal emphysematous destruction. 3 Although COPD is primarily a disease of the lungs and airways, it also has several extrapulmonary manifestations and comorbidities, such as cachexia, skeletal muscle abnormalities, osteoporosis, depression, anemia and cardiovascular disease. 3
Among the risk factors for developing COPD, both genetic and environmental factors are included, resulting in a complex, multifactorial and probably multigenic disease. 4 The environmental risk factor most frequently and clearly associated with COPD is cigarette smoking, with a prevalence of smokers who develop COPD ranging from 15–20% to 50% in elderly individuals.5,6 Nonetheless, estimates based on Global Initiative for Lung Disease spirometric criteria suggest that 17–38.8% of patients with COPD worldwide are non-smokers. 7 It is accepted that one of the most important risk factors for developing COPD in these subjects is biomass smoke (BS) exposure, as around 3 billion people worldwide are exposed to this pollutant. 8 Although the use of biomass fuels for cooking and heating purposes is more widespread in developing countries, an increasing number of households in developed countries also rely on biomass burning as a source of energy, owing to its cheapness, availability and near CO2 neutrality. 9
Solid biomass covers a wide range of materials from wood, straws and agricultural residues to animal dung or charcoal. The burning of these materials has the potential to release many airborne pollutants, including methane and volatile organic compounds, nitrogen oxides, sulfur oxides, hydrogen chloride, polyaromatic hydrocarbons, furans and dioxins, as well as organic and inorganic aerosol particulates, 10 known as particulate matter (PM). The respirable fraction of PM is classified according to its aerodynamic diameter into PM10 (diameter of ≤10 µm), PM2.5 (diameter of ≤2.5 µm) and PM0.1 (diameter of ≤0.1 µm). The composition of these PM is variable, although usually includes aeroallergens like pollen and other biologics such as fungal or bacterial elements, i.e. endotoxin. 11
Main common harmful components in both biomass and tobacco smoke.

BS enhances lung inflammation
COPD is considered an ‘archetypical disease of innate immunity’, 23 where inhaled particles and gases trigger an inflammatory response, favoring tissue proliferation in small airways and tissue destruction in lung parenchyma, in addition to the recruitment of immune cells to these compartments. 24 A considerable amount of evidence, from in vivo and in vitro studies, shows the potential of BS as an enhancer of this pulmonary inflammation.
Studies of BS exposure in animals provide the first line of evidence. A study by Fidan et al. reported severe histopathological effects in lungs of rabbits exposed to dried dung smoke, such as respiratory epithelial proliferation, alveoli destruction and increased emphysematous change scores. 25 Similar results were observed in rats exposed to BS. Thus, rats exposed to dried dung smoke also manifested higher levels of perivascular and peribronchial inflammation, as well as parenchymal infiltration, 26 while an increased number of inflammatory cells in bronchoalveolar lavage fluid (BALF) was observed in rats exposed to grain crust or wood smoke.27,28 In a murine study, Mehra et al. demonstrated that mice exposed to dung smoke had increased lung macrophages. 29 Moreover, these mice exhibited more perivascular inflammation and had higher granulocyte colony stimulating factor (G-CSF) and granulocyte-macrophage colony stimulating factor (GM-CSF) BALF levels than those exposed identically to cigarette smoke. In another study, instillation of ultra-fine carbon black particles into the lungs of mice also triggered higher numbers of pulmonary macrophages, neutrophils and higher levels of TNF-α in BALF when compared with controls. 30 In the same vein, Sussan et al. reported higher pro-inflammatory cytokine production, neutrophilic inflammation, airway resistance and hyper-responsiveness in mice acutely exposed to PM from cow dung compared with those exposed to PM from wood, whereas the latter showed higher eosinophilic inflammation in subchronic exposures. 31
Several in vitro studies have also shown the pro-inflammatory effects that BS exerts over innate immune cells. Thus, the epithelial type II pneumocyte cell line A549 showed an increased production of monocyte chemoattractant protein (MCP)-1 and IL-8 after exposure to PM from coal or wood smoke.32–34 Similarly, ambient air pollution PM10 particles induced the production of IL-6, IL-8, IL-1β, GM-CF and TNF-α in human bronchial epithelial cell lines.35,36 In turn, Williams et al. reported an enhancement in RNA transcript expression of the IL-1α, IL-1β, IL-6, IL-12, TNF-α, IFN-α, IFN-β and IFN-γ genes in CD14+ monocyte-derived dendritic cells stimulated with PM with a median diameter of 1.8 µm. 37 In another study, Alfaro-Moreno et al. showed that endothelial cells exposed to PM10 exhibited increases in the levels of TNF-α and GC-CSF, while no increase in pro-inflammatory mediators were observed in a mast cell line (HMC-1) under the same stimulation. 38 Interestingly, when this mast cell line was co-cultured with a macrophage cell line (THP-1) and stimulated with PM10, large increases were observed for the levels of IL-6, IL-8, G-CSF (>10-fold), MIP-1a and MIP-1b, IL-1β (up to 8-fold) and TNF-α (up to 100-fold). Indeed, alveolar macrophages (AMs), considered key orchestrators of immune responses in lungs, are one of most reactive cell types when exposed to inhaled pollutants, and BS is not an exception. In this sense, different studies reported an increased production of Il-1β, IL-6, IL-8, MIP-1, MIP-2, TNF-α and GM-CSF in cultured macrophages after exposure to wood smoke or PM.35,36,39–42 Furthermore, even cultured human lung fibroblasts have proven to be stimulated after exposure to wood smoke extract, showing an increased release of IL-8 and higher deposition of perlecan and fibronectin, two important extracellular matrix components that could contribute to the characteristic airway fibrosis in COPD. 43
Finally, some studies performed in patients with and without COPD are also consistent with the idea that BS fosters an inflammatory response in the lungs. Thus, increased pulmonary numbers of macrophages, neutrophils, eosinophils, mast cells and lymphocytes, as well as higher sputum levels of IL-6 and TNF-α, were reported in women exposed to biomass smoke when compared with those who used other types of fuel.44–46 Banerjee et al. also reported an increased expression of surface receptors involved in adhesion to endothelia and transmigration in circulating neutrophils of biomass users, 46 which could explain the increased extravasation of inflammatory cells to the lung of BS-exposed patients. In another study, BS-exposed patients with COPD showed significantly higher levels of neutrophils, eosinophils and IL-8 in induced sputum compared with healthy individuals. 47 Moreover, patients exposed to BS have an increased expression and activity of MMPs—involved in the turnover of extracellular matrix molecules and related to the pathogenesis of COPD—not only in lungs, but also in blood.47–49 This underlines that the pulmonary inflammatory response triggered by BS also has a systemic effect. Consistent with this, increases in CD8+ lymphocytes, NK cells, IL-6, IL-8, TNF-α, C-reactive protein and MCP-1 have been reported in the blood of BS-exposed individuals.49–52
The aforementioned studies describe acute effects of BS on immune cells; however, COPD is characterized by a chronic inflammatory pattern that continues even after the removal of the original noxious insults. While no study has yet assessed the pulmonary infiltration and/or activation of inflammatory cells after the BS exposure cessation, ongoing inflammation in patients with COPD that quit smoking has been reported.53–56 The reason why this inflammatory pattern persists remains a mystery, but most hypotheses point to a dysfunction affecting both innate and adaptive immune cells. 57
BS alters pulmonary host defense mechanisms
Several epidemiological studies showed increased risks of acute respiratory infections (ARI) associated with BS exposure,58–62 a matter of interest as ARI stand out as one of the leading causes of mortality, especially among children, in developing countries. 63 Moreover, some studies reported an association between tuberculosis cases and biomass fuel smoke exposure compared with exposure to cleaner forms of fuels. 64
Despite the link between chronic exposure to BS and the occurrence of respiratory infections being based primarily on epidemiological data, few experimental studies in animals have reinforced this idea. Thus, Hatch et al. showed a significantly increased mortality on mice with streptococcal pulmonary infection instilled with carbonaceous particles. 65 Additionally, a higher susceptibility to infections with Staphylococcus aureus in rats exposed to wood smoke in the range of 3 h–2 wk was also reported. 66 Furthermore, mice exposed to nitrogen dioxide, a main component of BS, showed an increased mortality from aerosolized Klebsiella and a reduced pulmonary antibacterial defense against S. cursive.67,68
Mechanisms of innate immunity modulation by BS
Collectively, the above mentioned studies provide evidence about the effect of BS as both an enhancer of lung inflammation and an impairer of pulmonary antimicrobial defense. A growing body of research has focused on shedding light on the mechanisms by which this pollutant alters the innate immune response. In this respect, two main classes of cell surface receptors—the PRRs and the transient receptor potential (TRP) channels—have shown the ability of transducing signals initiated after BS exposure (Figure 1).
Modulation of innate immune responses by biomass smoke in COPD. Three main classes of cell surface receptors, TLRs, SRs and TRPs channels have shown the ability of transducing signals initiated after BS exposure. The binding of certain components of BS, such as silica, to SR-A receptors seems to trigger an attenuation of the inflammatory response by decreasing the levels of pro-inflammatory mediators and activating apoptosis in alveolar macrophages. This could lead to an impaired immune response leading to the characteristic exacerbations in patients with COPD. BS is also capable of exerting two different responses via TLR4. On the one hand, it has been reported that PM could activate the pro-inflammatory transcription factors AP-1 and NF-kB through TLR4 binding. On the other hand, some studies have shown that PM down-regulates TLR expression and impairs macrophage activity. Both responses have been associated with emphysema and airway limitation. Finally, some components present in BS, such as PMs, acrolein, crotonaldehyde or zinc, elicit a pro-inflammatory response by signaling through the TRPA1 receptor. In addition to the exogenous agonists, the ROS generated during the inflammation could also activate TRPA1, thus contributing to the amplification of the immune response that is characteristic in COPD.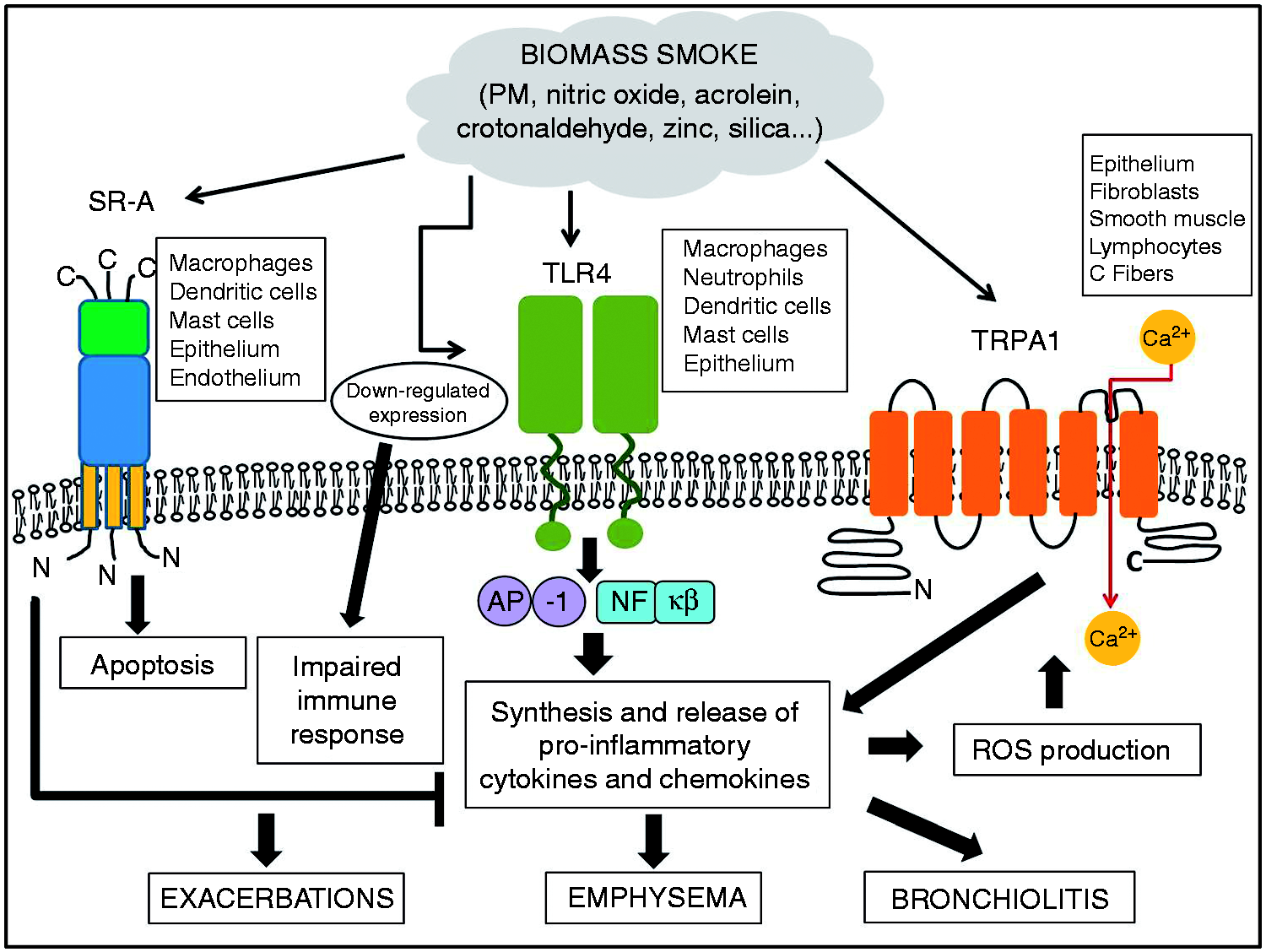
BS activates PRRs
PRRs are crucial in detecting PAMPs, such as bacterial and fungal cell wall components and viral nucleic acids. Detection of PAMPs by PRRs induces inflammatory responses and activates innate host defenses. In addition to representing an important defense against pathogens, among the several classes of known PRRs, TLRs and the class A scavenger receptor (SR-A) have also been shown to recognize elements of BS.
TLRs are key components of the innate defenses, having both endogenous and exogenous ligands. TLRs are predominantly expressed by monocytes/macrophages and neutrophils, but they are also expressed by mast cells, dendritic cells, NK cells, fibroblasts, airway epithelium, pneumocytes and even lung T cells. There are 13 TLRs described, of which TLR2 and TLR4 have been proved to bind PM. 69 It is furthermore accepted that the microbial components of PM are responsible for TLR activation after BS exposure.70–72 Hence, it has been proposed that BS may initiate pulmonary signaling through PM–TLR2/TLR4 binding, thus activating NF-кB and activator protein (AP)-1 pathways, and leading to the synthesis and release of pro-inflammatory cytokines and chemokines (Figure 1). 73 Consistent with this, it has been shown that both antagonist blocking and gene deletion of TLR2 and TLR4 inhibit the pro-inflammatory response of pulmonary cells to PM.36,74–77 However, and in spite of this pro-inflammatory effect through the TLR pathway, it has been shown that BS actually down-regulates the expression of these receptors. Thus, Becker et al. showed a decreased expression of TLR4 mRNA in cultured human AMs exposed to coarse and fine PM. 36 Sahlander et al. found a decreased expression of TLR2 on blood monocytes of non-smoking pig farmers exposed to organic material on a daily basis when compared with non-farming smokers and controls who underwent a bronchial LPS challenge. 78 In addition, Williams et al. showed a marked down-regulation of TLR2 and TLR4 in human myeloid dendritic cells exposed to ambient PM. 37 These results are in the same line as those describing an impaired phagocytosis and lymphocyte activation activity in wood smoke-exposed macrophages, suggesting that BS exerts something more than just a pro-inflammatory stimulus on these cells.69,79,80 The notion of a dysregulation in either TLR expression or in macrophage activity is of particular interest in COPD. Recent studies have shown that down-regulation or deficiency in TLR4 expression is associated with emphysema and airflow limitation in smokers,81,82 while impaired TLR responses in AMs correlate with propensity for COPD exacerbations. 83
Scavenger receptors (SRs) are another class of PRRs that are able to bind a diverse array of endogenous and foreign molecules. Although little is known about the immunological effects of BS through SRs, it has been reported that SR-A recognizes components of PM. 84 SR-A is predominantly expressed by macrophages, dendritic cells and mast cells, as well as epithelial and endothelial cells. 85 Intriguingly, unlike the pro-inflammatory response initiated after PM–TLR2/TLR4 binding, the activation of SR-A by BS components seems to trigger an attenuation of inflammation (Figure 1). Thus, SR-A2−/− mice exposed to silica, a main component in the ash of herbaceous crops and residues, 10 showed increased levels of TNF-α, mRNA levels of chemokine (C-X-C motif) ligand (CXCL)3 and a significantly increased neutrophilia when compared with wild type mice.86,87 Moreover, it has been shown that specific blocking of SR-A2 prevents PM10-induced apoptosis in AMs. 88 As COPD is characterized by a decreased apoptosis of AMs, 89 which contributes to maintain the chronic inflammatory response, a hypothetical reduction of SR-A expression in AMs of patients with COPD would not be surprising. Accordingly, Ganesan et al. reported an absence of SR-A expression in AMs in an animal model of COPD, involving mice infected with non-typeable Haemophilus influenzae, an important bacterial pathogen associated with COPD exacerbations. 90 Furthermore, the authors suggested that the lack of SR-A could be responsible for the observed defect in bacterial phagocytosis in these mice. Currently, no study assessing SR-A expression in human samples of patients with COPD has been reported, and further research is also needed in order to elucidate the mechanisms of BS through SR pathways.
BS activates TRPs
Recent attention has been paid to the TRP channels as mediators of innate immune responses triggered by BS. TRP channels—of which 28 members have been identified in mammals—are a family of cation-selective ion channels that can be activated by numerous chemical ligands (both exogenous and endogenous) or physical stimuli, hence acting as cellular sensors capable of responding to environmental changes. 91 These TRPs are subdivided into seven subfamilies: TRPC, TRPV, TRPM, TRPP, TRPML, TRPA and TRPN. Although there are some structural differences between TRP subfamily members, these receptors have six transmembrane segments—forming a pore between S5 and S6—and intracellular N- and C- termini. 92 Many of these TRPs exhibit Ca2+ permeability and are thus considered important regulators of the intracellular concentrations of this ion. This fact confers to TRPs an enormous potential to influence many cellular processes involved in lung diseases, as an increase of intracellular Ca2+ concentration is a powerful stimulus to the activation of virtually all pulmonary cell types.
Both the high capacity of TRPs to respond to a wide range of environmental irritants and the importance of smoking habit as a risk factor for lung disease have led to a growing body of studies assessing the role of several cigarette smoke components as TRPs agonists. 93 Although fewer studies have focused on BS as a TRP activator, many of the cigarette smoke components recognized by TRPs, e.g. nitrogen dioxide, acrolein, crotonaldehyde and zinc, are also found in BS (Table 1). Therefore, similar effects on TRP activation could be expected after cigarette smoke or BS exposure. Beyond the well-known consequences of TRP activation in airway sensory nerves, leading to coughing and neurogenic inflammation, 94 the idea that these receptors play a key role in regulating other inflammatory mechanisms is growing stronger (Figure 1). Indeed, TRPs are expressed not only in most cells of innate immunity such as macrophages, neutrophils, mast cells, eosinophils or epithelium, but also in endothelium, fibroblasts, neurons, smooth muscle cells and lymphocytes. 95 Several effects on immune responses have been described depending on the type of TRP and the cell studied.
Among all TRP subtypes, the TRP ankyrin 1 (TRPA1) and the TRP vanilloid 1 (TRPV1) are probably the most important in mediating immune responses after BS exposure. TRPA1 has been proved to be a molecular sensor for wood smoke PM in human alveolar cells. 96 Nassini et al. showed a release of IL-8 in cultured human airway epithelial cells, smooth muscle cells and fibroblasts exposed to acrolein; interestingly, this response was reduced by TRPA1 antagonists. 97 Moreover, an increase of CXCL-1 was found in BAL of wild type mice after intratracheal instillation of acrolein, an effect significantly reduced after pretreatment with a TRPA1 antagonist and completely absent in TRPA1−/− mice. Similar results were found by Mukhopadhyay et al. in cultured human lung fibroblasts and alveolar epithelial cells, showing an increased release of IL-8 in these cells when supplemented with crotonaldehyde and zinc; again, this response was attenuated by TRPA1-selective antagonists. 98 For their part, Deering-Rice et al. reported a coal ash-induced release of IL-6 and IL-8 in airway epithelial cells, which was inhibited by TRPV1 antagonists. 99 In the same study, the authors showed that instillation of coal ash into wild type mice lungs led to an increased expression of IL-6, as well as CXCL-1 and CXCL-2 mRNA in small airways, while this effect was attenuated in TRPV1−/− mice. Moreover, it has been suggested that TRPV1 may mediate PM-induced apoptosis. 100 Thus, it has been reported that airborne PM-induced apoptosis in human airway epithelial cells is prevented by capsazepine, a TRPV1 antagonist; 101 however, in bronchial human epithelial (BEAS-2B) and A549 cells, TRPV1 agonists have been reported to cause endoplasmic reticulum stress and cell death. 102
In addition to TRPA1 and TRPV1, there is evidence supporting a role for other TRP subtypes in pulmonary pathology and modulation of lung immune responses, although their association with BS is yet to be defined. For instance, Zhu et al. have reported seven TRPV4 single nucleotide polymorphisms associated with susceptibility to COPD. 103 Moreover, AMs of patients with COPD have a higher expression of TRP canonical 6 (TRPC6) mRNA compared with control subjects, suggesting a possible role of these receptors in the pathogenesis of the disease. 104 In a murine model of lung inflammation, Tiruppathi et al. showed a reduced plasma leakage in TRPC4−/− mice, underlining the importance of this TRP as a regulator of pulmonary microvascular permeability. 105 In another murine study, mast cells from TRP melastatin 4 knock-out mice (TRPM4−/−) showed an enhanced release of histamine, IL-6 and TNF-α compared with TRPM4+/+ cells, 106 while activation of TRPM8 in lung cells showed an enhanced expression of IL-1α and IL-1β, IL-4, IL-6, IL-8 and IL-13, as well as TNF-α and GM-CSF.107,108
Conclusions
The increasing prevalence of COPD, which is already among the five leading causes of death worldwide, is of global concern. Although no hypothesis so far can fully explain the pathogenic mechanisms of the disease, it is accepted that an abnormal innate immune response is involved. Furthermore, the paradigm of cigarette smoking as the main risk factor for developing COPD is beginning to shift towards BS exposure, as around 3 billion people worldwide are exposed to this pollutant daily. Certainly, BS has proven to be a noxious agent involved in the pathophysiology of several cardiorespiratory diseases, although the mechanisms of its detrimental effects on human health have just started to be uncovered. Both in vivo and in vitro studies have underlined the role of surface cell receptors such as PRRs and TRPs as mediators of lung innate immune modulation after cigarette smoke exposure. Although cigarette smoke and BS have many components in common, more research focusing specifically on the effects of BS is necessary in order to clarify its actual contribution to pulmonary disease. Moreover, the immune modulation of this pollutant depends on the type of cell surface receptor activated, ranging from a pro-inflammatory response to an increase of apoptosis in inflammatory cells. Therefore, following the advancement in the understanding of TRLs, SRs and TRPs as sensors of inhaled pollutants, future research should test their potential role as therapeutic targets in respiratory disorders such as COPD.
Footnotes
Acknowledgments
We would like to thank Dr. E. Brown for reviewing this article, as well as Dr. T.S. Ward and Miss S. Scher for linguistic advice.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Dr. Olloquequi is funded by the Chilean National Science and Technology Fund (CONICYT), FONDECYT Project N° 11150022.