
Abstract
The endothelium forms a vast network that dynamically regulates vascular barrier function, coagulation pathways and vasomotor tone. Microvascular endothelial cells are uniquely situated to play key roles during infection and injury, owing to their widespread distribution throughout the body and their constant interaction with circulating blood. While not viewed as classical immune cells, endothelial cells express innate immune receptors, including the Toll-like receptors (TLRs), which activate intracellular inflammatory pathways mediated through NF-κB and the MAP kinases. TLR agonists, including LPS and bacterial lipopeptides, directly upregulate microvascular endothelial cell expression of inflammatory mediators. Intriguingly, TLR activation also modulates microvascular endothelial cell permeability and the expression of coagulation pathway intermediaries. Microvascular thrombi have been hypothesized to trap microorganisms thereby limiting the spread of infection. However, dysregulated activation of endothelial inflammatory pathways is also believed to lead to coagulopathy and increased vascular permeability, which together promote sepsis-induced organ failure. This article reviews vascular endothelial cell innate immune pathways mediated through the TLRs as they pertain to sepsis, highlighting links between TLRs and coagulation and permeability pathways, and their role in healthy and pathologic responses to infection and sepsis.
Introduction
Endothelial cells line the inner surface of blood vessels and capillary beds, and serve as the interface between circulating blood and surrounding tissues. The average human adult is estimated to contain over 1 trillion endothelial cells that cover a surface area in excess of 1 million cm2 and altogether weigh approximately 1 kg.1–4 To put this vast number of endothelial cells in perspective, the average human adult is estimated to have roughly 20–50 billion peripheral blood mononuclear cells (PBMCs) and 1–4 billion monocytes circulating in their bloodstream.
5
Endothelial cells dynamically regulate the vascular barrier, modulate vasomotor tone, play central roles in coagulation and hemostasis, and are critically involved in the movement of leukocytes between the bloodstream and extravascular tissues (Figure 1). Endothelial cells are also increasingly being recognized as being key active participants in the host’s innate immune response to infection and injury.6,7
Major outputs after endothelial cell TLR pathways are activated. Endothelial cell TLRs engage microbial- and host-derived factors present within the vascular lumen to initiate inflammation. Activated endothelial cells (1) secrete cytokines and chemokines and express adhesion molecules that facilitate leukocyte movement between the blood and tissues at sites of infection and injury; (2) dysregulate coagulation homeostasis by activating the tissue factor (TF) pathway, causing increased levels of factors that induce thrombosis, and reduced levels of factors involved in clot breakdown (fibrinolysis); and (3) increase permeability of the endothelium which results in the movement of plasma fluid and proteins into tissues.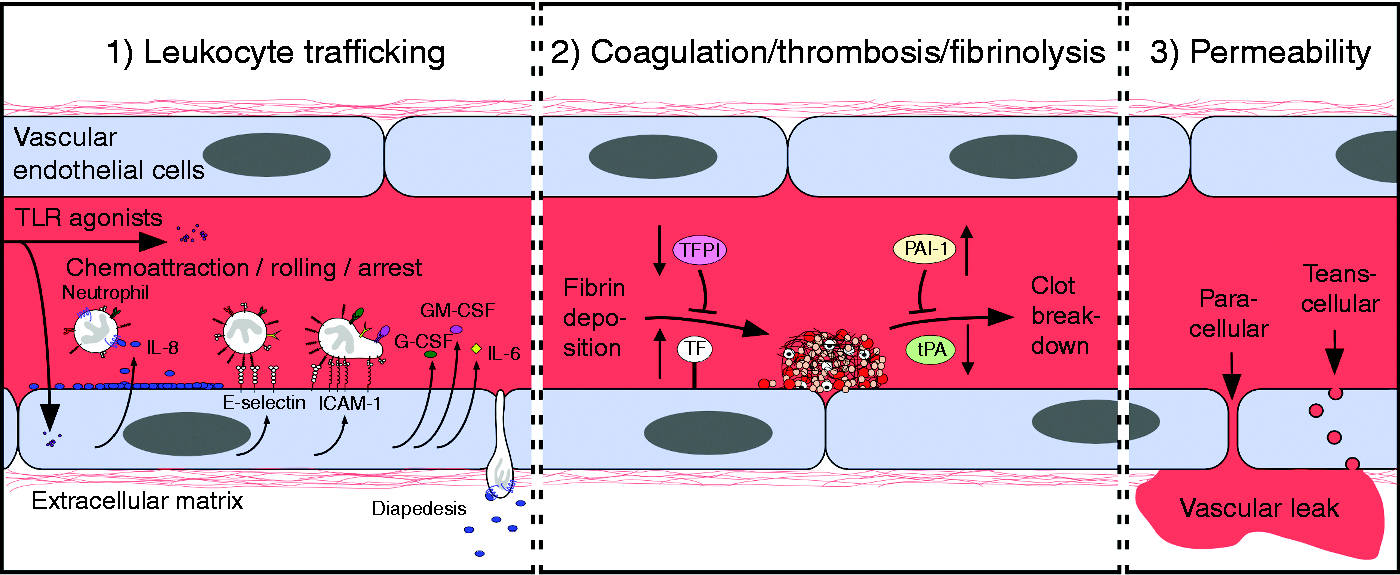
Whereas a great deal is known about the effects of microorganisms on monocyte and macrophage inflammatory pathways, substantially less is known about their effects on endothelial cells, which have not classically been viewed as immune cells. However, similar to leukocytes, endothelial cells express innate immune receptors, including members of the Toll-like receptor (TLR) family (Figure 2; Table 1),8–38 as well as NOD-like receptors and RIG-I like receptors.39–42 Engagement of endothelial innate immune receptors with microbial and host-derived agonists upregulates the expression of specific cytokines, chemokines and adhesion molecules, and increases the binding of neutrophils to the endothelium.6,10,11,14,17,18,20,26,30–32,34,43–50 The direct activation of endothelial innate immune pathways by TLR2, TLR4 and TLR9 agonists have been reported to modulate endothelial pathways involved in permeability and coagulation.8,16,22,26,30,38,46,51–57 The combination of inflammation, activation of coagulation pathways and increased vascular permeability may serve to create a physical barrier that limits the spread of infection into the bloodstream. The hypothetical concept of ‘hemostatic containment’ postulates that leukocyte adhesion to vessel walls and microvascular thrombosis directly obstruct vessels draining sites of infection, and that tissue edema resulting from increased vascular permeability further limits blood outflow by externally compressing vessels.58,59 In contrast to these putative beneficial functions, the dysregulated activation of endothelial inflammatory pathways leads to pathologic coagulopathy with diffuse microvascular thrombosis, increased leukocyte activation within multiple organs, and capillary leak leading to intravascular hypovolemia and edema. These latter dysfunctional endothelial responses are believed to promote the life-threatening syndromes of septic shock and sepsis-induced multiple organ failure.
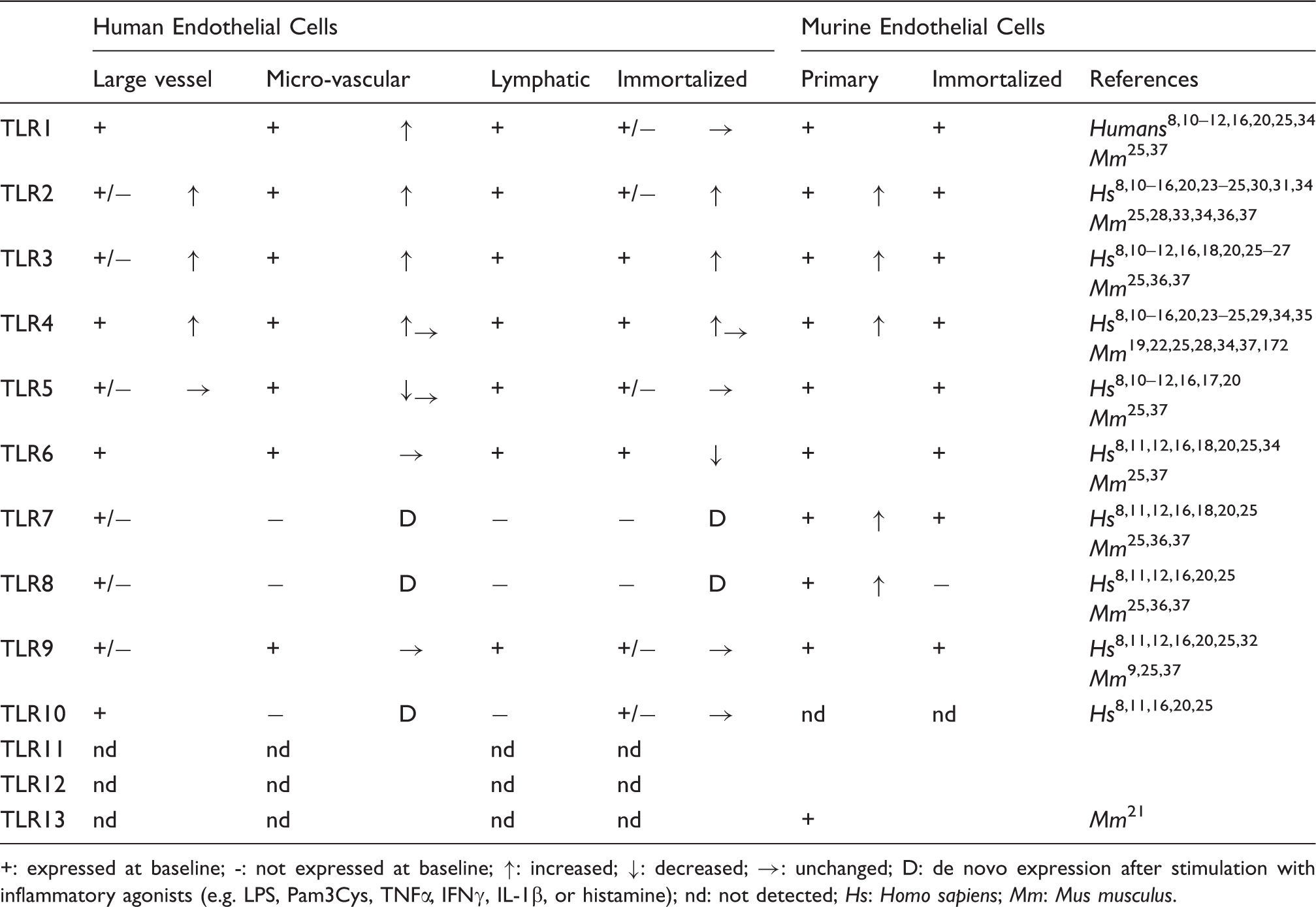
Vascular endothelial cell TLR pathways. After inflammatory agonists bind their cognate TLR receptors on endothelial cells, TLR dimers associate with MyD88 and/or TRIF adaptor proteins and activate downstream signaling via MAPK family members (i.e. p38, JNK and ERK5) and NF-κB to modulate pathways involved in inflammation, coagulation and vascular permeability. Of note, ERK5 has recently been shown to promote TLR signaling outcomes in endothelial cells, while ERK1/2 differentially regulates TLR signaling outcomes in endothelial cells and leukocytes. TLR expression in different endothelial cell types. +: expressed at baseline; -: not expressed at baseline; ↑: increased; ↓: decreased; →: unchanged; D: de novo expression after stimulation with inflammatory agonists (e.g. LPS, Pam3Cys, TNFα, IFNγ, IL-1β, or histamine); nd: not detected; Hs: Homo sapiens; Mm: Mus musculus.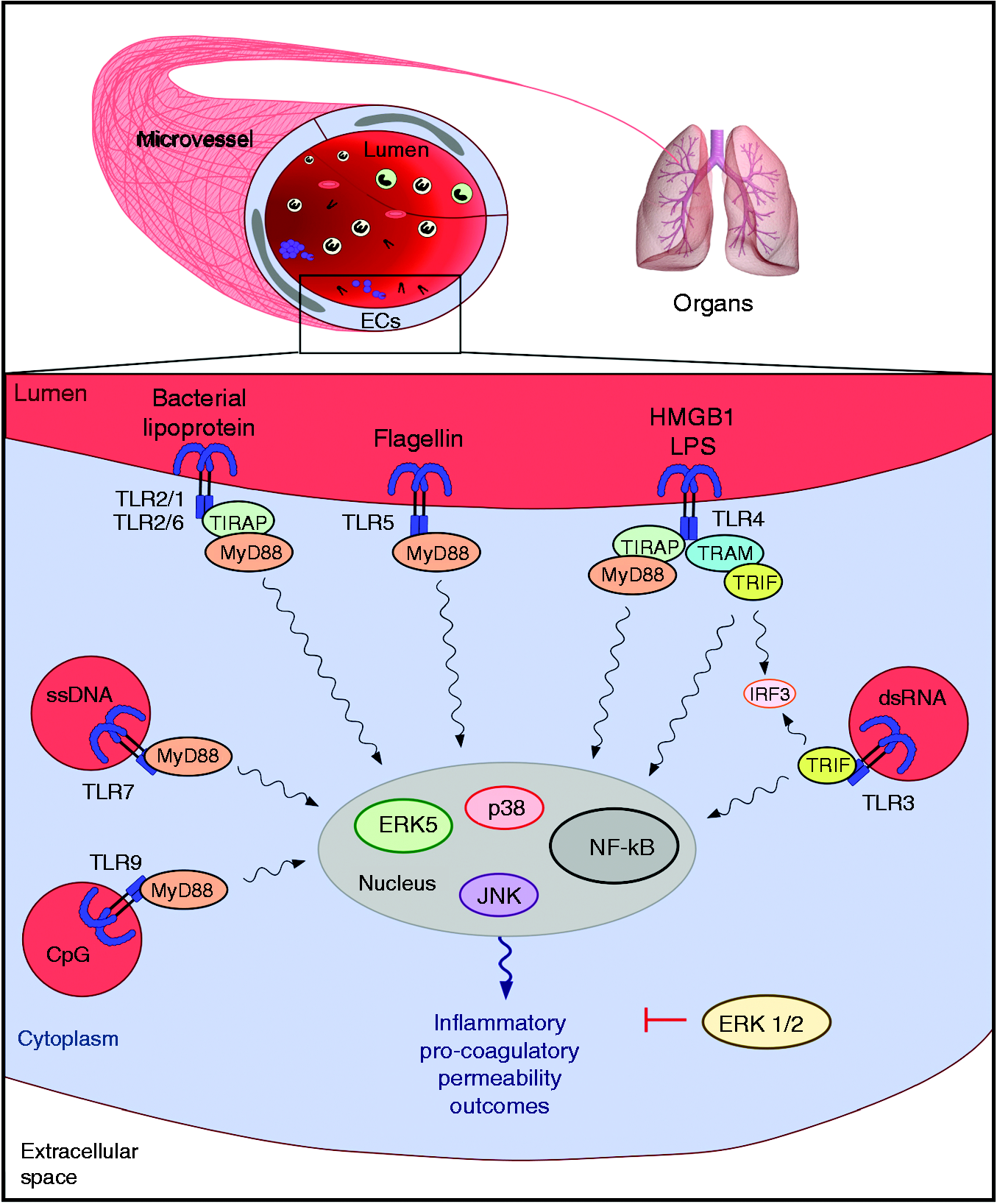
Responses of endothelial cells and leukocytes to TLR stimulation.
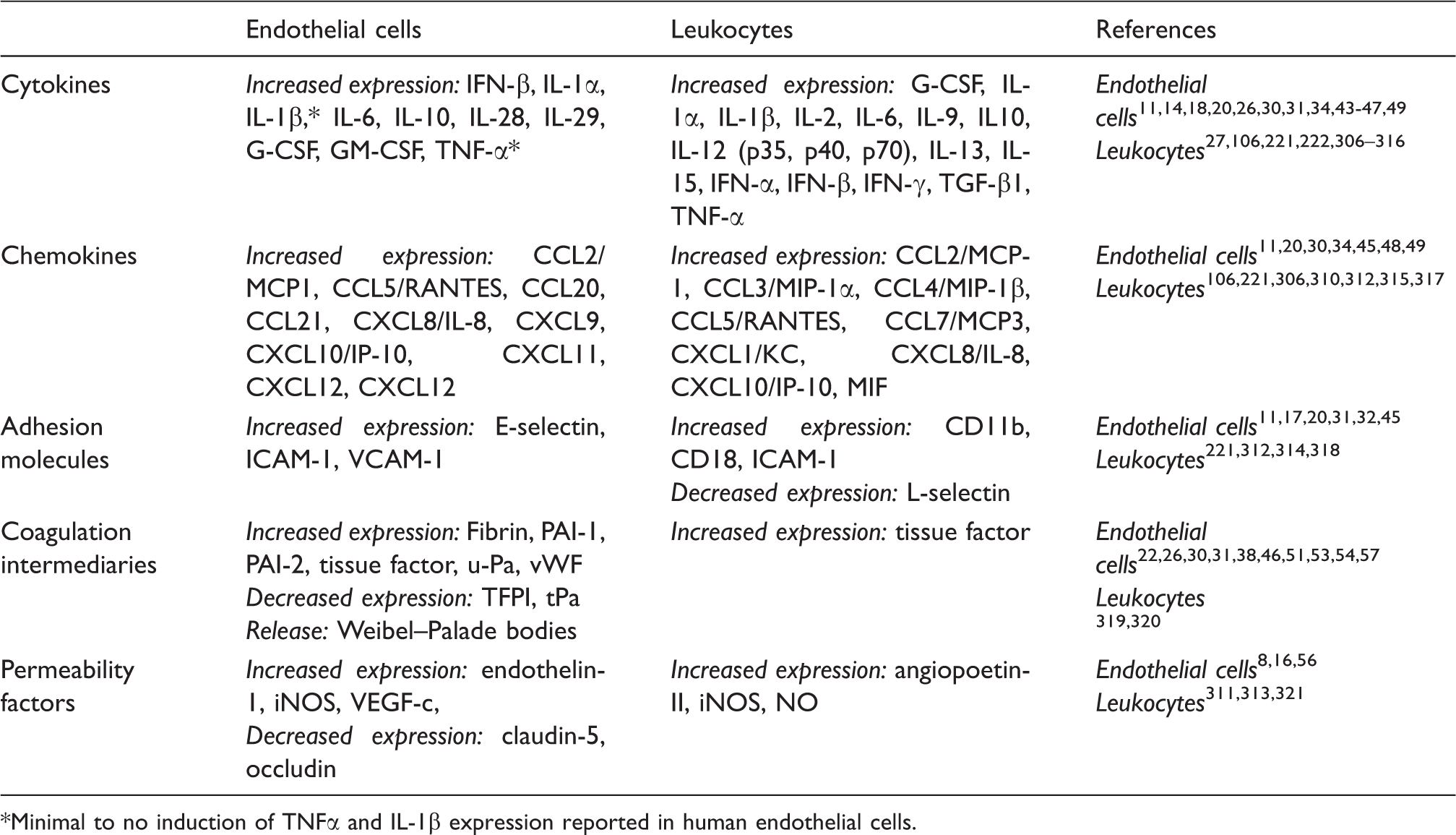
Minimal to no induction of TNFα and IL-1β expression reported in human endothelial cells.
Herein we review vascular endothelial innate immune pathways as they pertain to sepsis. We will focus on endothelial cell TLRs, intracellular signaling pathways, normal and dysregulated endothelial inflammatory responses, and immunomodulatory pathways in the endothelium.
Endothelial innate immune pathways in sepsis-induced endothelial dysfunction
Sepsis remains a major healthcare problem worldwide, with continued high morbidity and mortality. 64 Currently, sepsis is treated with antimicrobial agents, localized control of the source of infection with surgery or drainage, and the supportive care of dysfunctional or failing systems and organs. 65 With the recent negative outcomes of several Phase 3 clinical trials, there are currently no approved sepsis-directed adjuvant therapies.64,66–68 Although roughly half of patients who succumb to sepsis die of multiple organ failure, the mechanisms underlying sepsis-induced organ failure have yet to be fully unraveled, which is a barrier to developing effective sepsis-directed therapies.69,70 Endothelial dysregulation, coagulopathy with microvascular thrombosis, excessive vascular leak and increased leukocyte activation in organs lie at the heart of tissue injury and organ failure during sepsis. These interconnected processes occur within the microvasculature, and involve multiple cell types.
Microvascular endothelial cells are critically involved in the pathogenesis of sepsis-induced organ failure by virtue of their participation in coagulation, the vascular barrier, and neutrophil activation and trafficking.71,72 Because of their ubiquitous distribution throughout all vascular beds, endothelial cells participate in early and delayed inflammatory responses in sepsis. During the early phases of sepsis, endothelial cells are directly activated by microbial factors such as LPS and bacterial lipoproteins. Subsequently, endogenous inflammatory agonists that are newly synthesized or released by activated leukocytes and endothelial cells or by injured cells, such as IL-1β, TNFα, HMGB1, NO and pro-inflammatory prostanoids, further promote endothelial dysfunction, either on their own or in synergy with circulating microbial TLR agonists.
Sepsis-induced dysregulation of the balance of coagulation and fibrinolysis (clot breakdown) (Figure 1), leads to coagulopathy and, at the extreme, to the development of disseminated intravascular coagulation (DIC). 73 Even in the absence of overt DIC, diffuse microvascular thrombosis is believed to reduce blood flow and to promote organ injury. Despite recent negative human trials using recombinant activated protein C and tissue factor pathway inhibitor the pathologic coagulopathy of sepsis remains a focus of research on development sepsis therapeutics.67,74 Higher plasma levels of plasminogen activator inhibitor-1 (PAI-1) correlate with increased rates of organ failure and death in patients with sepsis. 75 PAI-1 inhibits fibrinolysis, thereby facilitating the persistence of microvascular thrombi, and the pro-coagulants tissue factor and thrombin are activated in sepsis. The direct activation of endothelial TLR2, TLR4 and TLR9 all modulate endothelial expression of coagulation pathway mediators in a pattern that is consistent with that observed in humans with sepsis (Table 2), suggesting that the activation of endothelial TLRs promotes sepsis-induced coagulopathy.22,30,38,46,51–54,57
Sepsis causes increased endothelial permeability, leading to fluid and protein leakage across the vascular wall with resultant intravascular hypovolemia with reduced tissue perfusion, as well as tissue edema. The mechanisms by which sepsis induces vascular permeability are complex, and most likely involve both the activation of endothelial cells by microbial TLR agonists and by host inflammatory mediators such as TNF-α.8,16,30,55,76,77 Studies suggest that activated neutrophils also independently promote endothelial dysfunction and contribute to barrier dysfunction.78–80 The direct activation of endothelial TLR2 or TLR4 by bacterial lipopeptides or LPS, respectively, increases endothelial permeability.8,16,30,55 In contrast, TLR9 agonists induce endothelial permeability in a neutrophil-dependent fashion. 81 Together, and perhaps synergistically, host and microbial inflammatory agonists induce paracellular permeability.55,76 LPS has been reported to TLR4-dependently induce paracellular permeability of human lung microvascular endothelial cells via TRAF6 and SRC family kinase-mediated tyrosine phosphorylation of zonula adherens proteins, including VE-cadherin, γ-catenin and p120ctn. 55
As outlined above endothelial cells actively contribute to the pathophysiology of sepsis. The constellation of intravascular hypovolemia, microvascular thrombosis and vasodilatation, tissue edema and extravasation of activated neutrophils into tissues leads to organ injury and ultimately multiple organ failure. Some still unanswered fundamental questions about the endothelium in sepsis include whether endothelial innate immune pathway activation directly promotes, or conversely protects against, sepsis-induced multiple organ failure, and the relative and integrated roles of endothelial and leukocyte innate immune pathways in sepsis outcomes.
Innate immune pathways and the healthy endothelial response to infection
Long underappreciated is that endothelial cells play a key role in sensing microorganisms and promoting early innate immune responses in sepsis.19,82 Because of their location, endothelial cells are among the first cell types to be exposed to circulating microbes and microbial toxins such as LPS. Indeed, the activation of endothelial TLRs upregulates endothelial cell secretion of cytokines and chemokines, including IL-6, CXCL8/IL-8 and CCL2/MCP-1, and upregulates endothelial expression of adhesion molecules, such as E/P-selectin, vascular cell adhesion molecule (VCAM)-1, and intercellular adhesion molecule (ICAM)-1, which facilitate leukocyte adherence to the endothelium and promote transendothelial migration into surrounding tissues (Figure 1; Table 2).6,30,49,50 Upon their release from endothelial cells, cytokines play key roles in leukocyte production, activation and survival.49,83,84 Furthermore, secreted chemokines, such as IL-8 and CCL2 form intravascular gradients on the surface of the endothelium near the site of inflammation that direct neutrophils and inflammatory monocytes, respectively, to the site of infection or injury.85–87 In addition to acting as chemoattractants, IL-8 and CCL2 also activate signaling pathways in leukocytes that lead to conformational changes of β2 - and α4-containing integrins to higher affinity states, which further promotes their adherence to the endothelium.88–90
At sites of endothelial cell activation, E- and P-selectins mediate leukocyte capture and rolling through relatively weak associations with molecules such as P-selectin glycoprotein 1 (PSGL1) and L-selectin present on the leukocyte cell surface. These associations also increase integrin affinity for its substrate, which facilitates a more robust attachment of leukocytes and their eventual arrest on the endothelium, despite the shear forces of ongoing blood flow.91–93 Resident/patrolling monocytes utilize the integrin LFA-1/αLβ2 and CX3CR1 to crawl along the endothelium, while inflammatory monocytes arrest via an association between the integrin VLA-4/α4β1 on their surface and VCAM-1 on the surface of activated endothelium.94–97 Neutrophils, however, arrest via a stable association between the integrins LFA-1/αLβ2 or Mac-1/αMβ2 on their surface and ICAM-1. 7 Additionally, neutrophil and endothelial sialidase activity has been reported to desialylate the endothelial cell surface, which leads to hyperadhesiveness of neutrophils to the endothelium and facilitates neutrophil diapedesis. This sialidase-mediated desialylation is independent of protein upregulation or the adhesion molecules, and is believed to cause the rapid and early amplification of neutrophil-mediated host responses.98,99 After their arrest along the vascular endothelium, leukocytes migrate out of the vasculature to sites of infection and inflammation using para- or transcellular routes. 100
Once leukocytes extravasate to infected or injured tissues, they can eliminate pathogens or necrotic cellular debris through phagocytosis. 101 Neutrophils, which are the first leukocytes to accumulate in sites of infection, generate neutrophil extracellular traps (NETs), which directly bind, kill and prevent the spread of invading microorganisms.102–104 Neutrophils can also release chemokines directed at the recruitment of other leukocytes, including macrophages and dendritic cells (DCs), such as CCL4, alarmins, such as cathepsin G, the antimicrobial peptide LL-37 and defensins.102,105 Because of their specialized ability to clear pathogens and/or present antigens to lymphocytes, leukocytes that are activated by TLR stimulation express a distinct series of effector molecules, dominated by cytokines such as IL-12p70, in order to initiate activation of the adaptive immune system (Table 2).106,107
In addition to their role in leukocyte activation and trafficking, endothelial cells also respond to infections and sterile injury by regulating hemostasis. 108 Hemostasis is a finely tuned balance of coagulation, anticoagulation and fibrinolysis. The activation of endothelial TLRs causes a shift toward a pro-coagulant/pro-thrombotic state that promotes fibrin clot formation and reduces clot breakdown (Figure 1).30,109 Strong links between inflammation and coagulation are evolutionarily conserved, suggesting a survival advantage to this linkage.58,59 For example, horseshoe crabs (Limulus polyphemus) are ancient arthropods that have an open circulatory system, with the hemocyte as their only blood element. 110 When activated by pathogens, Limulus hemocytes induce clotting which sequesters an infected area from the remainder of its body, thereby containing the infection. The link between inflammation and clotting is the basis of the Limulus amoebocyte lysate (LAL) assay that is used to quantify LPS. 111 Similarly to the ancient horseshoe crab, activation of coagulation may serve a beneficial role in humans by promoting the formation and persistence of thrombi, which help to physically contain a microbial threat by trapping microbes in the fibrin clots themselves.112–117 In addition, activation of coagulation may facilitate antibacterial defenses, as suggested by reports that the binding of leukocytes to fibrin-immobilized pathogens via integrin Mac-1/αMβ2 promotes bacterial clearance.118,119 Furthermore, coagulation factors can act as chemotactic agents and the presence of thrombi restricts blood flow, which together can increase the likelihood of recruiting circulating leukocytes to inflamed tissues.120–123
Because of their proximity to smooth muscle cells, endothelial cells also control vasomotor tone. 124 After TLR activation, endothelial cells increase the expression of vasodilators, such as NO (Table 2), which decreases blood pressure and has variable effects on tissue blood flow. 125 The vasodilation and concurrent downregulation of junctional proteins cause the expansion of the gaps between endothelial cells, which allows fluids and small molecules to leak into the extravascular compartment.126–128 The increased endothelial permeability also enhances the ability of leukocytes to adhere to the endothelium and extravasate into surrounding tissues to initiate immune activation and repair pathways.126,129–131
Importantly, these primary effects of endothelial TLR activation work in interconnected feedback loops that themselves can amplify endothelial cell inflammatory responses. For example, cytokines released by endothelial cells can regulate the expression of coagulation factors, and coagulation factors can, in turn, modulate the expression of cytokines and adhesion molecules.132–136 Factors involved in coagulation and fibrinolysis, such as PAI-1 and tissue factor, also affect vascular permeability pathways.137–139 Thus, the inflammatory pathways of endothelial cells are intricately structured and are crucial to the host’s response to infection.
Endothelial cells as targets and reservoirs of bacteria
While endothelial cells are known targets of several bacteria, including Porphyromonas gingivalis, Rickettsia rickettsii, Staphylococcus aureus and Bartonella,140–143 limited work has focused on understanding mechanisms by which endothelial cells themselves clear intracellular bacteria. Murine endothelial cells have been reported to clear Rickettsia conorii via the NO production and the induction of autophagy. 144 Studies with HUVECs suggest that the clearance of Rickettsia via NO is dependent upon the specific cytokine environment of the endothelium and requires the production of hydrogen peroxide. 145 Although these reports indicate that endothelial cells are capable of targeting and degrading intracellular pathogens, intracellular survival of S. aureus has been reported in human endothelial cells.146–148 In fact, endothelial cells that are infected with S. aureus are believed to serve as a reservoir for recurrent S. aureus bacteremia.146,149 In particular, small colony variants of S. aureus can survive inside of endothelial cells for prolonged periods, and persist intracellularly during infection.150,151 These latter studies with S. aureus support the basic hypothesis that microorganisms dwell quiescently in endothelial cells evading host immune responses, and causing recurrent bacteremia and the development of metastatic tissue foci of infection.
Endothelial cell TLRs
The TLRs were first identified based on their sequence homology with the Drosophila Toll protein. Toll was originally characterized as a gene necessary for proper development of dorsal–ventral polarity in the Drosophila embryo, but subsequently was found to be indispensible for the antifungal immune response.152–155 To date, 10 TLRs have been identified in humans (TLRs 1–10) and 12 in mice (TLRs 1–9 and 11–13). Tlr10 appears nonfunctional in a variety of mouse strains, owing to a retroviral insertion, but may be functional in rats, while the gene encoding TLR11 in humans contains premature stop codons, and orthologs for Tlr12 and Tlr13 have not been identified in EST database screens.156–158 Most of the TLRs form homodimers, with the exceptions of TLR2, which forms heterodimers with either TLR1 or TLR6 and TLR10, which has been reported to form heterodimers with TLR1 and TLR2.156,159 The TLRs are expressed in intracellular compartments or at the cell surface in a variety of cell types and each member binds a conserved subset of PAMPs derived from viral [i.e. dsRNA (TLR3) and ssRNA (TLR7/8)], fungal [i.e. zymosan (TLR2)] or bacterial [i.e. lipoproteins (TLR1/2/6), LPS (TLR4), flagellin (TLR5) and CpG-DNA (TLR9)] pathogens.160–170 TLR2, TLR4 and TLR9 also bind several endogenous damage-associated molecule patterns (DAMPs) such as heat-shock proteins, hyaluronan and versican, HMGB1 and heparin sulfate or mitochondrial DNA. 171 Binding of TLR agonists to their cognate TLR induces TLR dimerization, and activation of downstream signaling pathways that can lead to substantially different outcomes depending on the cell type. Here we will focus on TLR2, TLR4 and TLR9, which have been most extensively studied in endothelial cells and are the principal TLRs that are known to play important roles in bacterial sepsis.
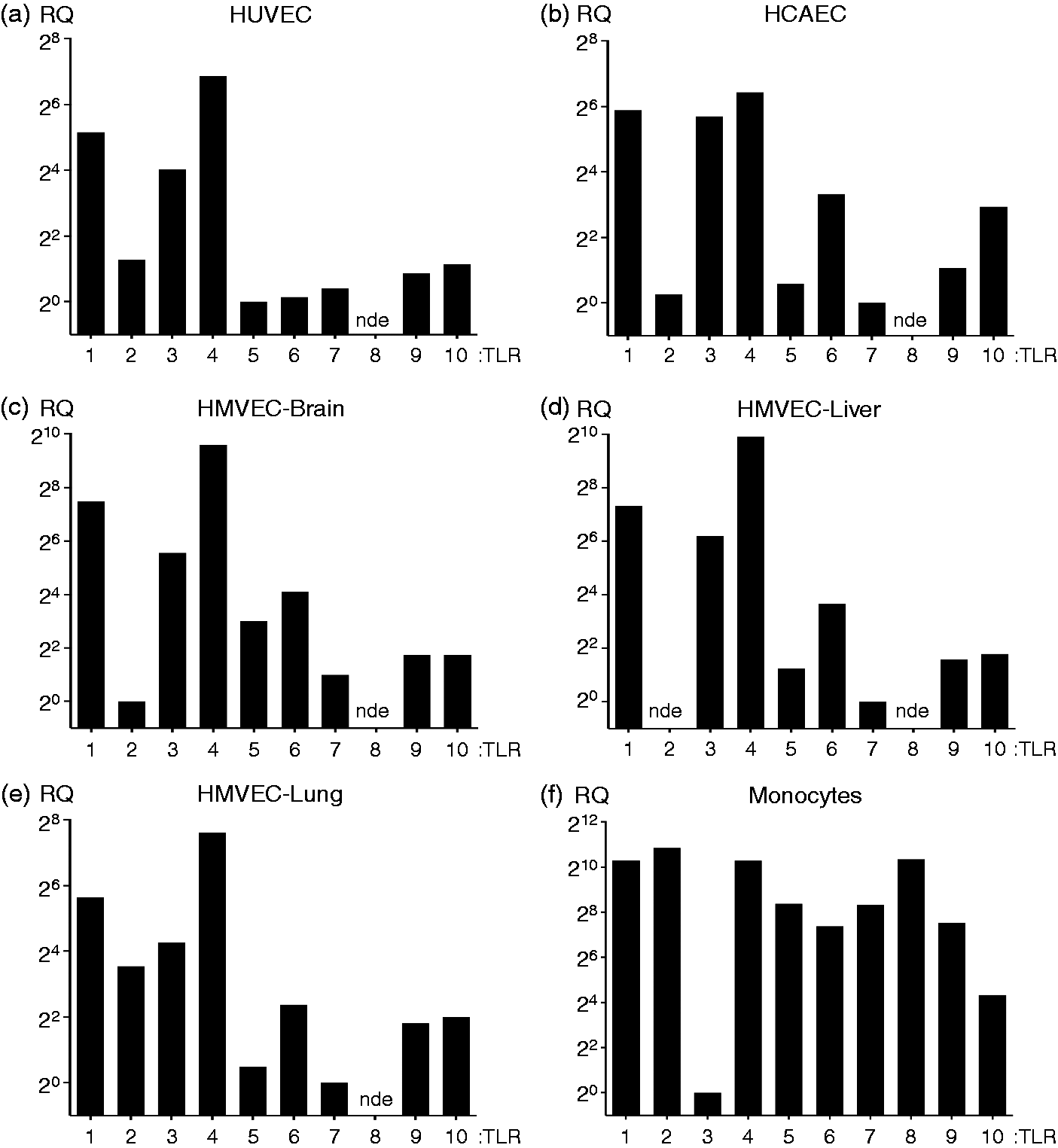
Endothelial cells have been reported to express all of the TLRs with the possible exception of TLR8 (Table 1).8–37,172 Compared with leukocytes endothelial cells express relatively low levels of most of the TLRs at baseline, except for TLR3 and TLR4, which are robustly expressed in HUVECs and human microvascular endothelial cells (HMVECs) from multiple vascular beds. Studies suggest that in contrast to their surface expression in monocytes, in endothelial cells TLR2 and TLR4 are also localized intracellularly.31,173 Figure 3 shows data from our laboratory on the expression of TLR transcripts in different endothelial cells, including HUVEC, human coronary artery endothelial cells (HCAEC), HMVEC-brain, HMVEC-liver and HMVEC-lung, as well as in primary human monocytes, which we have included to demonstrate the relative levels of the different TLRs in endothelial cells and monocytes. Although TLR2 is expressed at low levels at baseline, we and others have found that treatment of human endothelial cells with microbial and host factors (i.e. histamine, HMGB1, Pam3Cys, MALP-2, TNFα, IFNγ, LPS and IL-1β) induces a robust upregulation of TLR2.14,15,28,31,33,173–175 This induction of TLR2 expression in endothelial cells has not been observed in human leukocytes,
176
which suggests that the transcription of TLR2 is regulated differently in human endothelial cells and leukocytes.
Human endothelial cells from different endothelial cell niches express comparable levels of TLR mRNA transcripts. Human cells were cultured, lysed and mRNA isolated in order to compare the relative expression of TLR gene transcripts in different endothelial cell populations compared with human monocytes at baseline. (A) HUVEC (passage 2, multiple donors; Lonza), (B) HCAEC (passage 4, female donor; Lonza, Walkersviille, Maryland, US), (C) HMVEC-brain (passage 4, unknown donor sex; Cell Sciences, Canton, Massachusetts, US), (D) HMVEC-liver (passage 4, unknown donor gender; Cell Sciences), (E) HMVEC-lung (passage 4, two female donors; Lonza), were grown in either EGM-2 or EGM-2 MV media (Lonza), incubated at 37℃ under humidified 5% CO2 and lysed with Trizol (Invitrogen, Carlsbad, CA, USA) upon confluency. (F) Primary human monocytes were isolated from PBMCs by gradient centrifugation using Lymphoprep (Axis-Shield, Oslo, Norway) followed by purification on magnetic columns using MACS CD14+ beads (Miltenyi Biotech Inc, San Diego, California, US), and immediately lysed in Trizol. Heparinized whole blood was collected by venipuncture from a healthy human volunteer. Three biological replicates were analyzed for each donor. Specific gene expression assays and the manufacturer’s suggested assay reagents were purchased from Applied Biosystems (Foster City, CA, USA). mRNA concentrations were determined with a ND-1000 (NanoDrop;Thermo Fisher Scientific, Grand Island, New York, US) and mRNA was reverse transcribed to cDNA using the High Capacity RNA-to-cDNA Kit using 2 µg of mRNA per reaction (Invitrogen). An input of 10 ng cDNA in 10 μl total reaction volume per well containing TaqMan Fast Advanced Master Mix (Applied Biosystems) was used in all qPCR experiments and qPCR was performed using the StepOnePlus System (Applied Biosystems). Run method: PCR activation at 95℃ for 20 s was followed by 40 cycles of 1 s at 95℃ and 20 s at 60℃. The mean Ct value for HPRT1 and GUSB was used as the reference in calculating the ΔCt values for each biological replicate. The data analysis was performed using the 2 -ΔΔCt method; however, for multiple donor assays, the data were corrected using log transformation, mean centering and auto scaling to ensure appropriate scaling between biological replicates.322,323 The relative quantification (RQ) values shown in the graphs are relative to lowest detectable expressing TLR gene for each cell type. When gene expression was not detected in more than half of the technical replicates, it was defined as not expressed (nde: not detectably expressed). The methods of calculation utilized assume an amplification efficiency of 100% between successive cycles. Note: in our previously published manuscript, TLR expression analysis in HUVEC was performed using 2.5 ng cDNA, and therefore the lowest expressing TLRs were not detected.
31
Despite reasonably low baseline TLR2 expression, bacterial lipopeptide and lipoprotein TLR2 agonists strongly activate human endothelial cells. For example, treatment with bacterial lipopeptides TLR2-dependently activates NF-κB and induces the phosphorylation of p38 MAPK, JNK and ERK5 at 30 min in primary human lung HMVECs and HUVECs, and upregulates tissue factor levels in HUVEC lysates as early as 1 h.30,31 We hypothesize that endothelial cells dynamically express TLR2 in order to calibrate the host’s response to different levels of infectious threats or injury. Because TLR2 recognizes a broad range of microbial and host inflammatory agonists, the low basal expression of TLR2 may serve to limit excessive systemic endothelial activation during minor infections or the transient episodes of bacteremia that occur during normal daily life when there is a break in mucosal or skin integrity. The upregulation of endothelial TLR2 after exposure to inflammatory agonists may promote a more robust endothelial inflammatory response that serves both to facilitate and augment leukocyte responses, and regulate endothelial permeability and localized coagulation pathways. Furthermore, while TLR2 is primarily localized at the plasma membrane in leukocytes, we have reported the intracellular localization of TLR2 in primary human endothelial cells. 31 This suggests that similar to TLR4 and TLR9 agonists, bacterial lipopeptides may have to be first internalized to activate endothelial TLR2.32,173 Alternately, intracellularly localized TLR2 may be necessary to recognize intracellular pathogens.140,177,178
In addition to being critical in inflammatory responses to microbial factors, activation of TLRs by endogenous inflammatory agonists exacerbates endothelial dysfunction and promotes sepsis-induced organ injury. Endogenous inflammatory agonists are released by activated leukocytes and endothelial cells, and by cells that have undergone ischemia reperfusion (IR) injury resulting from the tissue hypoperfusion. IR injury complicates sepsis, as well as a variety of sterile inflammatory processes that also cause organ injury such as trauma, hemorrhage, cardiac arrest and organ transplantation. TLR2, TLR4 and TLR9 have each been implicated in IR injury, and endogenous TLR4 agonists have been reported to circulate in sepsis and other forms of IR injury.179–188 The specific role of endothelial TLR4 in sensing endogenous mediators and promoting sepsis-induced organ injury has not been clearly defined, but multiple host factors, including HMGB1, hyaluronan, HSP70 and S100A8 have been found to circulate in sepsis and TLR4-dependently induce the inflammatory activation of endothelial cells.189–193 These studies support the widely held belief that the activation of TLR4 by circulating host mediators serves to generate, amplify and/or perpetuate inflammation, and to promote sepsis-induced organ failure.
Endothelial TLR-dependent signaling
The majority of research on TLR signaling pathways is derived from studies using leukocytes. In both leukocytes and endothelial cells, homodimerization of TLR4 or TLR9, and heterodimerization of TLR2 with either TLR1 or TLR6, initiates intracellular signaling cascades that ultimately lead to the activation of the MAPK family members, and the transcription factor NF-κB.194,195 TLR4 associates with the adapter protein MD2 in order to recognize LPS and initiate downstream signaling.196,197
TLR4 has been reported to function intracellularly in endothelial cells. 173 LPS binding protein catalyzes the formation of LPS–CD14 complexes and their uptake and delivery to intracellular TLR4–MD2. 173 This multiprotein complex, which includes soluble CD14 (sCD14), is required for the activation of endothelial cells by low concentrations of LPS (0.1–10.0 ng/ml). 173 CD14 is necessary for the activation of endothelial cells by low and moderate LPS concentrations, but it is not required for activation by high LPS concentrations (≥ 1 µg/ml). 198 While studies suggest that sCD14 may be relatively more important than membrane CD14 (mCD14) in endothelial activation by LPS, low-dose LPS has been reported to require both sCD14 and mCD14, suggesting that the two forms of CD14 differentially regulate LPS-induced activation of endothelial cells.173,198,199 Finally, while at high concentrations of LPS neither sCD14 nor mCD14 is required for the induction of E-selectin in endothelial cells, mCD14 is necessary for MyD88-independent activation of IFN-β. 198 Variability in the endothelial cell expression of CD14 between studies may have resulted from the loss of expression of CD14 with the passage of endothelial cells, as suggested by a report that CD14 expression becomes undetectable in HUVECs at passage three. 199
With the notable exception of TLR3, TLR signaling originates with the association of the cytosolic Toll-interleukin-1 receptor (TIR) domain of the TLRs and the TIR domain-containing adapter MyD88.200–204 TLR3 and TLR4 TIR domains can also associate with TIR-domain-containing adapter-inducing interferon-β (TRIF/TICAM1) to activate the transcription factor IRF3.101,205,206 TLR2 and TLR4 also utilize the adaptor TIR domain-containing adapter protein [TIRAP or MyD88-adaptor-like (MAL)], which is required to bridge MyD88 to the cytoplasmic domains of TLR2 and TLR4.206–210 The pathways that lead to the activation of MAPKs and NF-κB downstream of TLR2, TLR4 and TLR9 have been extensively described elsewhere for leukocytes.101,211,212 Notably endothelial cells, like leukocytes, express the intermediary TLR signaling components that are required for activation of the MAPKs and NF-κB.
NF-κB
NF-κB activity is centrally involved in inflammatory gene expression in endothelial cells, including those induced by TLR2, TLR4 and TLR9.9,15,31,32,50,213,214 NF-κB is also necessary for the upregulation of TLR2 observed in endothelial cells activated with bacterial lipoprotein or LPS.15,31 While TLR4 and TLR9 agonists strongly activate NF-κB in human endothelial cells, we found that TLR2 agonists induce a less robust activation of NF-κB, especially in comparison to human leukocytes. 31 Interestingly, however, the TLR2-induced activation of NF-κB has a prolonged duration, whereas in monocytes the kinetics of NF-κB activation is strong but relatively short. 31 Conceivably, these differences in NF-κB activation profiles in human endothelial cells and monocytes are responsible for the differences observed in downstream inflammatory outcomes in these two cell types.
MAPKs
The conventional MAPK family members include p38-MAPK (α, β, γ and δ), c-Jun N-terminal kinase (JNK; 1, 2 and 3), extracellular-signal-regulated kinase (ERK)-1/2 and ERK5. 215 These serine/threonine kinases are activated by upstream MAPK kinases (MAP2Ks), which are themselves activated by MAP2K kinases (MAP3Ks). The MAPKs, and their downstream MAPK-activated protein kinases (MAPKAPKs), can induce inflammatory gene expression by three distinct mechanisms: (1) directly influencing the activity of transcription factors, (2) regulating proteins involved in transcriptional and translational control, including chromatin reorganization, and (3) modulating the integrity of protein complexes.215–227 Furthermore, substantial crosstalk between the NF-κB and the MAPK pathways may exist in some cell types.228–234 Treatment with the TLR2 agonist Pam3Cys has been reported to activate p38-MAPK, JNK and ERK5, but not ERK1/2 in HUVECs, and p38-MAPK, JNK and ERK1/2 in HMVECs.31,235 Treatment with the TLR4 agonist LPS has been reported to activate p38-MAPK and JNK in HUVECs, and p38-MAPK, JNK and ERK1/2 in HMVECs.214,235,236 Finally, treatment with the TLR9 agonist CpG DNA has been reported to activate p38-MAPK, but not ERK1/2, in mouse lung endothelial cells. 9 Because of the importance of the conventional MAPKs in endothelial cell signaling, and because there are some apparent differences in the role of the ERKs in endothelial cells and leukocytes, each is reviewed in more detail below.
Both p38-MAPK and JNK have been studied extensively and play critical roles in multiple cellular processes, including inflammation. p38-MAPK can promote gene and protein expression in several ways: (1) regulating activator protein (AP)-1 transcription factors (e.g. members of the ATF, Jun and Fos families); (2) stabilizing mRNA; (3) influencing NF-κB activity; (4) modulating gene expression by regulating chromatin modifiers and remodelers; and (5) influencing protein translation.215,223,224,228,237–244 The primary role of JNK activation downstream of TLRs is to regulate transcription factors such as ATF family members (e.g. ATF2) and JUN family members (e.g. c-Jun). 215 In conjunction with c-Fos, these transcription factors form AP-1 heterodimers that promote inflammation. However, whether specific AP-1 dimers either positively or negatively regulate gene transcription is dependent upon may factors, including the proteins levels of the dimerization partners, the composition of the AP-1 dimers, various post-translational modifications and the availability of certain accessory proteins. 245
The role of ERK1/2 downstream of TLRs is complex. Evidence suggests that ERK1/2 generally promote pro-inflammatory responses in leukocytes but negatively regulate inflammation in endothelial cells.31,101,246 Studies to define the role of ERK1/2 have been hampered by the fact that several commonly used pharmacological inhibitors of MEK1, the upstream kinase of ERK1/2, inhibit the activity of both MEK1 and MEK5, the upstream kinase of ERK5 [e.g. PD98059, U0126 or PD184352 (> 1 µM)].247–251 Therefore, it is not always clear whether the observed outcomes are due to the loss of ERK1/2 or ERK5 kinase activity. We observe that inhibition of endothelial MEK1 (PD184352; < 1 µM) induces the upregulation of TLR2 even in the absence of TLR agonists, and augments TLR2-dependent expression of TLR2, PAI-1 and inflammatory proteins (i.e. IL-6, GM-CSF and G-CSF) in endothelial cells. 31 The mechanism by which MEK1 negatively regulates TLR2 signaling in endothelial cells is currently not known.
ERK5 is a recently identified mediator of the inflammatory response, but the signaling pathways that lead to the activation of ERK5 downstream of TLRs are not known. Several reports suggest ERK5 has an anti-inflammatory role,252–257 while others, including ours, have reported a pro-inflammatory role in a variety of cell types, including downstream of TLRs in endothelial cells.31,258–266 It is possible that blood flow conditions dictate whether ERK5 serves a pro- vs. anti-inflammatory role in endothelial cells. For example, under normal physiologic conditions where blood flow is laminar ERK5 protects against vascular inflammation, whereas during sepsis and tissue injury/distress where there are areas of decreased and non-laminar blood flow, ERK5 promotes inflammation. In support of this concept is the finding that the small ubiquitin-like modifier (SUMO)-ylation of ERK5 promotes inflammatory pathways under conditions of disrupted laminar flow, which points to the possibility that post-translational modifications of ERK5 may switch its function from anti- to pro-inflammatory. 253 Furthermore, ERK5 may downregulate NF-κB activity through the expression of Krüppel-like factor 2 (KLF2), which has been proposed to have anti-inflammatory properties in healthy endothelial cells under laminar flow but not in areas of compromised blood flow.267–272 Further study is required to fully understand the mechanism of ERK5 inflammatory pathway regulation downstream of endothelial TLRs.
Immune-modulating pathways in the endothelium
Upon TLR activation, negative feedback loops are activated that initially moderate the inflammatory response, and eventually return TLR signaling pathways to homeostasis. Many mediators and pathways that negatively regulate TLR signaling have been identified in myeloid cells but their role in negatively regulating endothelial TLR signaling has not been established. 273 Several mechanisms are plausible in endothelial cells, including (1) the degradation of TLR signaling intermediaries (e.g. via SOCS-1); (2) the disruption of protein associations (e.g. via A20 or other DUBs); (3) sequestration of TLR signaling components (e.g. via ST2); (4) the inactivation of the MAPKs [e.g. via dual specificity phosphatases (DUSPs)]; (5) the synthesis of inhibitory proteins (e.g. IkBs, RelB and IkBNS); or (6) the silencing of signaling molecules by microRNAs (e.g. miR-146 a).273–276 There is also accumulating evidence that ERK1/2 activates negative feedback pathways that help terminate innate immune signaling pathways. For example, in leukocytes, treatment with LPS activates MSK1/2 downstream of ERK1/2, which leads upregulated expression and activation of DUSP-1 (MSK-1), which negatively regulate the activation of both p38-MAPK and JNK.212,277–279 Interestingly, the treatment of macrophages deficient in either MSK1/2 or DUSP1 with various TLR agonists, including bacterial lipopeptide, results in an augmented expression of pro-inflammatory cytokines, which is similar to our observations in TLR2 agonist activated endothelial cells treated with MEK1/2 inhibitor.31,277,280 This suggests ERK1/2 may play an important role in negative feedback loops TLR signaling pathways in endothelial cells.
Maladaptive responses in sepsis and tissue injury are, in part, caused by a failure to shift to the pro-resolving phase of inflammation. It is now recognized that the resolution of inflammation is an active process that is promoted by pro-resolving lipid mediators. 281 The group of specialized pro-resolving lipid mediators (SPMs), including lipoxins, resolvins, protectins and maresins, reduce inflammation and restore homeostasis to an infected or injured area by preventing the influx of more neutrophils, stimulating efferocytosis, and promoting the clearance of cellular debris by resolving macrophages.281–283 SPMs promote inflammatory resolution by suppressing NF-κB activation, in part via PPAR-γ. 284 Because SPMs do not seem to compromise host defense, inducers of SPM pathways are being considered for inflammatory therapeutics. 281 Additionally, while some prostaglandins (PGs) are pro-inflammatory, others promote the resolution of inflammation. For example, PGD2 upregulates IL-10 release via the receptor D prostanoid receptor 1 (DP1).281,282,285 Furthermore, 15-deoxy-Δ12,14-PGJ2 (15 d-PGJ2), the non-enzymatic metabolite of PGD2, promotes inflammatory resolution through the direct inhibition of IkB kinase, and modification of the DNA-binding domains of NF-κB subunits, and similar to SPMs, via PPAR-γ mediated suppression of NF-κB activation.281,286–288
The majority of research has analyzed the effects of lipid mediators on leukocyte function, but there are indications that lipid mediators also promote the resolution of endothelial inflammation. For example, Resolvin D2 (RvD2) stimulates the production of prostacyclin and NO by endothelial cells, both of which have vasoprotective effects. 289 In fact RvD2 was recently found to promote the resolution of acute inflammation and organ protection through the G-protein coupled receptor GPR18, which is expressed in human vascular endothelial cells.290,291 Additionally, PGD2 and 15 d-PGJ2 have been shown to reduce the inflammatory activation of endothelial cells during the resolution phase of the inflammatory response.292–299 Finally, recent studies suggest that another group of arachidonic acid-based lipids, the endocannabinoids, also have immunomodulatory activity.290,300,301 The endocannabinoids are ligands for cannabinoid receptors 1 and 2 (CB1R and CB2R) and the transient receptor potential channel V1 (TRPV1). Activation of CB2R, which is robustly expressed by leukocytes, has been reported to dampen the inflammatory response to infection.302–305 Notably, human endothelial cells express CB1R and TRPV1 at baseline, and CB2R during acute inflammatory events. 290 We recently discovered that the endocannabinoid N-arachidonoyl dopamine (NADA) modulates the TLR2- and TLR4-dependent inflammatory activation of human microvascular endothelial cells via CB1R, CB2R and TRPV1. 290 We speculate that, similar to the SPMs, NADA may also downregulate endothelial inflammation via the PG pathways and reduced NF-κB activation. Together, these reports suggest that lipid mediators may be important, overlooked immunomodulators of endothelial cells that require further study.
Summary
Under optimal conditions the endothelium contributes to beneficial host responses during infection. However, under more pathologic conditions, the unbridled or dysregulated activation of endothelial inflammatory pathways causes profound endothelial dysfunction with consequent shock and organ failure. Endothelial cells regulate vascular homeostasis and are critically involved in the host’s response to sepsis and to the development of multiple organ failure. Endothelial cells express innate immune receptors and signaling pathways, and are directly activated by TLR agonists. Although endothelial cells have long been known to produce cytokines and chemokines, and to facilitate leukocyte trafficking to tissues, they are underappreciated as significant contributors to the host’s immune response to infection. Interestingly, cultured primary human endothelial cells and monocytes produce comparable levels of IL-6 and IL-8 per cell when exposed to TLR agonists such as LPS or bacterial lipopeptides.31,34,49,62,63 Given that the number of vascular endothelial cells vastly exceeds the number of circulating monocytes by an order of magnitude (1 billion circulating monocytes vs. >1 trillion endothelial cells), endothelial cells may represent a major source of IL-6, IL-8 and other inflammatory mediators in sepsis. This concept is supported by a report that TLR-dependent activation of endothelial cells is a major source of IL-8 during bacterial infection in mice. 306 Additionally, while there is overlap between TLR signaling pathways and outcomes in leukocytes and endothelial cells, fundamental differences exist. For example, endothelial TLR activation induces negligible or no upregulation of TNFα and IL-1β, whereas activation of monocyte or macrophage TLRs robustly upregulates both of these cytokines.49,60,61 Furthermore, endothelial TLR signaling pathways also regulate endothelial permeability, and induce PAI-1, tissue factor and E-selection expression, outcomes that are not observed in leukocytes (Table 2).
The available data implicate endothelial TLR-dependent pathways in the inflammatory response during sepsis and in the development of sepsis-induced organ inflammation. We speculate that endothelial innate immune pathways could be exploited therapeutically to alleviate the detrimental aspects of endothelial dysfunction. For instance, therapies could target endothelial intracellular innate immune pathways through endothelial-specific delivery of pathway inhibitors or by exploiting differences in leukocyte and endothelial innate immune pathways. Alternatively, or in addition, lipid immunomodulators such as the SPMs or endocannabinoids could be used to facilitate resolution of endothelial inflammation and restoration of homeostasis. The specific targeting of endothelial inflammatory pathways could potentially reduce the development of organ failure, without interfering with the role of leukocytes in innate immune defenses and in generating adaptive immune responses. However, because endothelial cells play critical roles in maintaining physiologic homeostasis and contribute to the inflammatory response in sepsis, it is possible that endothelial-based therapies would deleteriously affect the host’s immune and physiologic responses to sepsis. Thus, a more comprehensive understanding of the complex roles that endothelial cells play in both beneficial and harmful responses to sepsis will be necessary to determine whether or not endothelial cell pathways represent viable targets for sepsis-directed therapies.
Footnotes
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.