
Abstract
Autophagy represents a key pathway in innate immune defense to restrict Mycobacterium tuberculosis growth inside host macrophages. Induction of autophagy has been shown to promote mycobacterial phagosome acidification and acquisition of lysosomal hydrolases, resulting in the elimination of intracellular M. tuberculosis reference strains such as H37Rv. The notorious Beijing genotype has been previously shown to be hyper-virulent and associated with increased survival in host cells and a high mortality rate in animal models, but the underlying mechanism that renders this family to have such advantages remains unclear. We hypothesize that autophagic control against M. tuberculosis Beijing strains may be altered. Here, we discovered that the Beijing strains can resist autophagic killing by host cells compared with that of the reference strain H37Rv and a strain belonging to the East African Indian genotype. Moreover, we have determined a possible underlying mechanism and found that the greater ability to evade autophagic elimination possessed by the Beijing strains stems from their higher capacity to inhibit autophagolysosome biogenesis upon autophagy induction. In summary, a previously unrecognized ability of the M. tuberculosis Beijing strains to evade host autophagy was identified, which may have important implications for tuberculosis treatment, especially in regions prevalent by the Beijing genotype.
Introduction
Autophagy (macroautophagy) is an evolutionary conserved cellular homeostatic process for qualitative and quantitative control of cytoplasmic biomass by removing long-lived proteins, cytosolic toxic aggregates and defunct organelles.1–4 In addition, autophagy has been increasingly appreciated for its role as a primordial form of cellular innate immunity against invading pathogens.1,2,5 Autophagy can be initiated when cells encounter stresses such as nutritional and immune insults resulting from starvation, a change in the level of immune mediators, and the presence of structural components of microbes and their products.1–4 Upon responding to an upstream stress signal, Atg proteins are organized into complexes and collaborate to generate a specialized double-membrane autophagosome.3,4 These complexes include (i) the protein kinase complexes ULK1 and AMPK to activate Beclin-1, (ii) the class III phosphatidylinositol 3-kinase complexes VPS34 and Beclin-1 to generate PI3P for recruitment of downstream effectors, and (iii) the Atg5/Atg12/Atg16 conjugation complexes that act as an E3 ligase for LC3 lipidation required for elongation and closure of an autophagosome.1,3,4 Cytosolic substrates can be either non-selectively engulfed into an autophagosome or selectively recognized and collected into autophagosome by different types of cargo receptors/adaptors.6–8 Upon closure, an autophagosome fuses with the lysosome resulting in the delivery of lysosomal acid hydrolases, which then degrade the enclosed contents. The resulting digested molecules, such as amino acids, are then released and recycled back into the cytoplasm.1,3,4
Autophagy has been shown to play important roles in cells’ autonomous defense against intracellular bacteria, parasites and viruses.1,9–11 In the context of Mycobacterium tuberculosis infection, induction of autophagy by starvation or other autophagy inducers results in the killing of the tubercle bacilli by overcoming the block in phagolysosomal biogenesis, as demonstrated by the acidification and acquisition of lysosomal hydrolases into compartments harboring the mycobacteria,12–17 even when they have survived in phagocytes by evading immunological mechanisms. 18 The proposed mechanistic details for autophagy-mediated mycobacterial elimination include (i) the direct engulfment of mycobacteria into autophagosomes and subsequent delivery to lysosomes for degradation, and (ii) the generation of killing peptides converted from innocuous cytosolic substrates engulfed and partially digested in autophagosomes/autophagolysosomes and subsequent delivery of these killing peptides to mycobacteria via fusion of autophagic vacuoles with mycobacterial phagosomes. 19 The importance of the autophagy pathway in antimycobacterial defense was substantiated by the findings that mice deficient in Atg5 expression in macrophages display a high susceptibility to mycobacterial infection.20,21 Furthermore, autophagy was shown to be required for effective antimycobacterial drug action of isoniazid and pyrazinamide, two of the first-line tuberculosis drugs, in an animal model of tuberculosis. 22 In addition, a genome-wide siRNA screen to identify host factors that regulate mycobacterial load in macrophages infected with different strains of M. tuberculosis revealed autophagy as the main host cell functional module that is perturbed by the pathogen. 23 Moreover, polymorphisms in the human autophagy and autophagy-related genes IRGM and P2X7 are found to be associated with susceptibility to tuberculosis.24–28 All of these data underscore the importance of autophagic process in mycobacterial control and indicate that the magnitude of autophagic elimination of mycobacteria may be the key determinant of their virulence and pathogenesis.
Mycobacterium tuberculosis can be subdivided into several distinct genotypic families/lineages, 29 among which the Beijing genotype, regarded as the highly successful lineage of M. tuberculosis often associated with multidrug resistance, was shown to be globally distributed, representing around 50% of strains in East Asia and more than 13% of strains worldwide. 30 The success of the Beijing family was thought to stem from its associated hyper-virulent phenotypes, as demonstrated by their higher ability to survive in host macrophages, to cause higher bacterial load and mortality in animal models, and to cause heavy AFB smear-positive sputum in patients.31–36 However, the mechanism attributing to the greater ability of M. tuberculosis Beijing genotype to survive in host cells remains unclear. As autophagy has been shown to be important for controlling M. tuberculosis growth inside host cells but the characterization of this pathway has been conducted using M. tuberculosis laboratory reference strains such as H37Rv and the vaccine strain BCG,12–17,37 but not the clinical strains such as those belong to the Beijing genotype, we set out to determine whether there is an alteration in autophagic control against strains of the Beijing genotype which may explain the hyper-virulence possessed by this family. Our results showed that the Beijing strains display greater ability to resist autophagic killing than that of the reference strain H37Rv and a strain belonging to the East African Indian (EAI) genotype.29,38 In addition, we revealed a possible underlying mechanism in which the resistance of the Beijing strains to autophagic restriction is not simply achieved by blocking autophagy-mediated acidification of their phagosomes or by inhibiting the autophagic flux in host cells but achieved by preventing autophagolysosome biogenesis, as demonstrated by their ability to inhibit the increased acquisition of cathepsin D, an enzyme of lysosomal acid hydrolases and a marker of lysosome, into mycobacterial compartments upon the induction of autophagy.
Materials and methods
Cell and bacterial culture
Mouse macrophage RAW 264.7 cells (ATCC) were maintained in DMEM (GE Healthcare, South Logan, UT, USA), 10% FBS (GE Healthcare, South Logan, UT, USA) and 4 mM
Fluorescent dye, Abs, DNA construct and siRNAs
LysoTracker Red (LTR; Invitrogen, Carlsbad, CA, USA) was used at 1:4000. For immunofluorescence assays, monoclonal Ab against Cathepsin D (R&D Systems, Minneapolis, MN, USA) was used at 1:200. For immunoblotting, polyclonal Abs against Beclin-1 (Santa Cruz, Santa Cruz, CA, USA) were used at 1:500 and monoclonal Ab against Actin (Abcam, Cambridge, UK) was used at 1:2000. The plasmid construct used in this study was as previously described. 41 All siRNAs used in this study were from Dharmacon (Lafayette, CO, USA).
Macrophage transfection
RAW264.7 cells were transfected with 5 µg cDNAs or 1.5 µg siRNAs as previously described. 42 In brief, 10–15 × 106 cells were re-suspended in 100 µl of Nucleofector solution kit V (Amaxa, London, UK). Plasmid DNAs or siRNAs were then added to the cell suspension and cells were nucleoporated using the Amaxa Nucleofector apparatus with the program D-032. Cells were then transferred into a new flask containing 12 ml complete media and incubated at 37℃ followed by a medium change at 6 h of incubation. Cells were then used in assays after 24 h of transfection with plasmid DNAs or 48 h of transfection with siRNAs.
Macrophage infection, mycobacterial survival and immunofluorescence microscopy
Infection of RAW264.7 cells with mycobacteria and quantification of mycobacterial survival after autophagy induction were carried out as previously described. 15 In brief, 3 × 105 cells of RAW264.7 macrophages were plated onto each well of 12-well plates 12 h before infection. Cells were then infected with a single-cell suspension of mycobacteria in complete media at an MOI of 10:1 for 1 h. Cells were then washed three times with PBS to remove un-internalized mycobacteria and autophagy was then induced by treatment with starvation media for 4 h. Cells were then lyzed and survival of bacteria was determined by plating for CFU. For immunofluorescence microscopy, 3 × 105 Raw264.7 macrophages were plated onto coverslips in 12-well plates 12 h before infection. Cells were then infected with 3 × 106 Alexa 488- or Alexa 405-labeled mycobacteria per well in complete media at 37℃ for 15 min, washed three times in PBS and chased for 1 h in complete media as previously described. 42 Cells were then washed three times with PBS and autophagy was then induced by treatment with starvation media for 2 h. Cells were then fixed with 4% paraformaldehyde/PBS for 15 min followed by permeabilization with 0.1% Triton X-100/PBS for 5 min. Coverslips were then blocked in PBS containing 3% BSA and stained with primary Abs according to the manufacturer’s recommendation. Cells were washed three times with PBS and then incubated with appropriate secondary Abs (Invitrogen) for 1 h at room temperature (25℃). Coverslips were then mounted using ProLong Gold Antifade Mountant (Invitrogen) and analyzed by confocal microscopy using the Zeiss LSM-700 Laser Scanning Microscope (Carl Zeiss, Jena, Germany). For LTR staining, cells were prestained in complete media containing LTR for 2 h at 37℃ before infection. Subsequent steps were carried out as described above but in the presence of LTR until the cells were fixed. For analysis of RFP–GFP–LC3 transfected cells, cells were fixed and mounted as described above. The numbers of RFP+GFP+–LC3 puncta (autophagosomes) and RFP+GFP−–LC3 puncta (autolysosomes) were determined in cells that contained mycobacteria in infection conditions and compared them with those of the uninfected control. At least 50 phagosomes per experimental condition in three independent experiments were quantified. For quantification, the percentage mycobacteria–marker colocalization was a fraction of total mycobacterial phagosomes examined and counted as positive when one or more puncta were observed on or in contact with the mycobacterial phagosomes.
Immunoblotting
Cells were lysed in lysis buffer containing 20 mM Tris, 100 mM NaCl and 1% NP-40. Cell lysates were then separated by 10% SDS-PAGE and proteins were transferred onto a nitrocellulose membrane (Amersham Biosciences, Little Chalfont, UK). Membranes were then blocked with 5% blocking solution (Roche Diagnostics) for 1 h before incubation with appropriate primary Abs at 4℃ overnight (16 h). Membranes were then washed three times with PBS containing 0.1% Tween 20 (0.1% PBST) followed by incubation with appropriate HRP-conjugated secondary Ab (Pierce, Rockford, IL, USA) at room temperature for 1 h. Membranes were then washed four times with 0.1% PBST followed by incubation with a chemiluminescence substrate (Roche Diagnostics, Mannheim, Germany) at room temperature for 2 min. Proteins were then detected with the enhanced chemiluminescence method.
Statistical analysis
Unless indicated otherwise, all experiments were independently conducted at least three times and data were pooled for presentation as mean ± SEM. All data were analyzed with Prism software (GraphPad, La Jolla, CA, USA) using two-tailed unpaired Student’s t-tests. P-values <0.05 were considered significant.
Results
The Beijing strains resist autophagic killing of mycobacteria by host cells
It has been well documented that induction of autophagy by starvation or other autophagy inducers in host macrophages results in the killing of mycobacteria reference strains such as M. tuberculosis H37Rv and the vaccine strain BCG.12,14,16,43–45 However, the autophagic killing capacity of host cells against M. tuberculosis clinical strains has not been characterized in detail. Therefore, we set out to evaluate whether different M. tuberculosis strains possess differing abilities to alter host autophagic killing capacity. As a control, we observed no variation in the infection of RAW264.7 macrophages by different M. tuberculosis strains when compared with that of the reference strain H37Rv (Figure 1A). Our results showed that, similarly to what has previously been observed,14,15,37,46 induction of autophagy in RAW264.7 macrophages by starvation, a classical method to stimulate autophagy, for 4 h can efficiently kill intracellular M. tuberculosis reference strain H37Rv by around 50% (Figure 1B). In addition, autophagy induction in RAW264.7 macrophages is able to restrict comparably an M. tuberculosis strain belonging to the EAI genotype (Figure 1B). In contrast, autophagy induction by starvation cannot promote the elimination of the strains belonging to the Beijing genotype (BJY and BJN) (Figure 1B). It should be noted that the viability of cells infected with different strains of M. tuberculosis is > 90%. This is also consistent with previous studies that used an MOI of 10 of M. tuberculosis to infect RAW264.7 macrophages for short-term incubation (4 h).13–15,42,46 In addition, to confirm that the observed mycobacterial control is mediated through the autophagy pathway, we targeted Beclin-1 by siRNAs in RAW264.7 macrophages and determined M. tuberculosis survival by CFU analysis. Beclin-1 targeting was verified by immunoblotting (Figure 1C). Depletion of Beclin-1 resulted in a decrease in starvation-induced elimination of M. tuberculosis reference strain H37Rv and the EAI strain confirming the role of autophagy in the killing of these mycobacteria (Figure 1D). In addition, Beclin-1 knockdown does not alter the resistance of the Beijing strains to starvation-induced elimination (Figure 1D). Altogether, these results indicate that autophagic control capacity of host cells against different strains of M. tuberculosis is not identical and, unlike what has previously been observed with the laboratory reference strains, which are effectively killed by autophagy induction, the Beijing strains are able to resist autophagic restriction by host cells. The observed alteration in autophagic killing capacity against different strains of M. tuberculosis was not related to the presence of intact pks 15/1, a gene encoding a polyketide synthase required for the synthesis of phenolic glycolipid, a known virulence factor of mycobacteria,
47
as both the BJY (pks 15/1 present) and BJN (pks 15/1 absent) can similarly resist autophagic killing by host cells while the EAI (pks 15/1 present) is efficiently eliminated by autophagy induction (Figure 1B, D).
The Beijing strains of M. tuberculosis possess a greater ability to resist starvation-induced autophagic killing in host macrophages. (A) RAW264.7 macrophages were infected with different M. tuberculosis strains at an MOI of 10 for 1 h. After washing the cells multiple times to remove un-internalized mycobacteria, cells were then lyzed and the number of mycobacteria that infected host cells was determined by plating for CFU. (B) RAW264.7 macrophages were infected with different M. tuberculosis strains at MOI of 10 for 1 h. After washing the cells multiple times to remove un-internalized mycobacteria, cells were then induced to undergo autophagy by starvation for 4 h. Cells were then harvested and the number of viable mycobacteria was determined by plating for CFU. (C) RAW264.7 macrophages were transfected with siRNAs against Beclin-1 or scramble (Scb) control. After 48 h of transfection, RAW264.7 cells were collected and processed for immunoblot analysis to estimate the efficacy of Beclin-1 depletion. (D) RAW264.7 macrophages were transfected with siRNAs as in (C). At 48 h after transfection, cells were infected with different strains of M. tuberculosis and then subjected to autophagy induction by starvation and CFU analysis as in (B). Data are mean ± SEM from at least three independent experiments. **P < 0.01 and †P ≥ 0.05, all relative to the full control set to 100%. EAI, the East African Indian strain with intact pks15/1; BJY, the Beijing strain with intact pks15/1; BJN, the Beijing strain without intact pks15/1.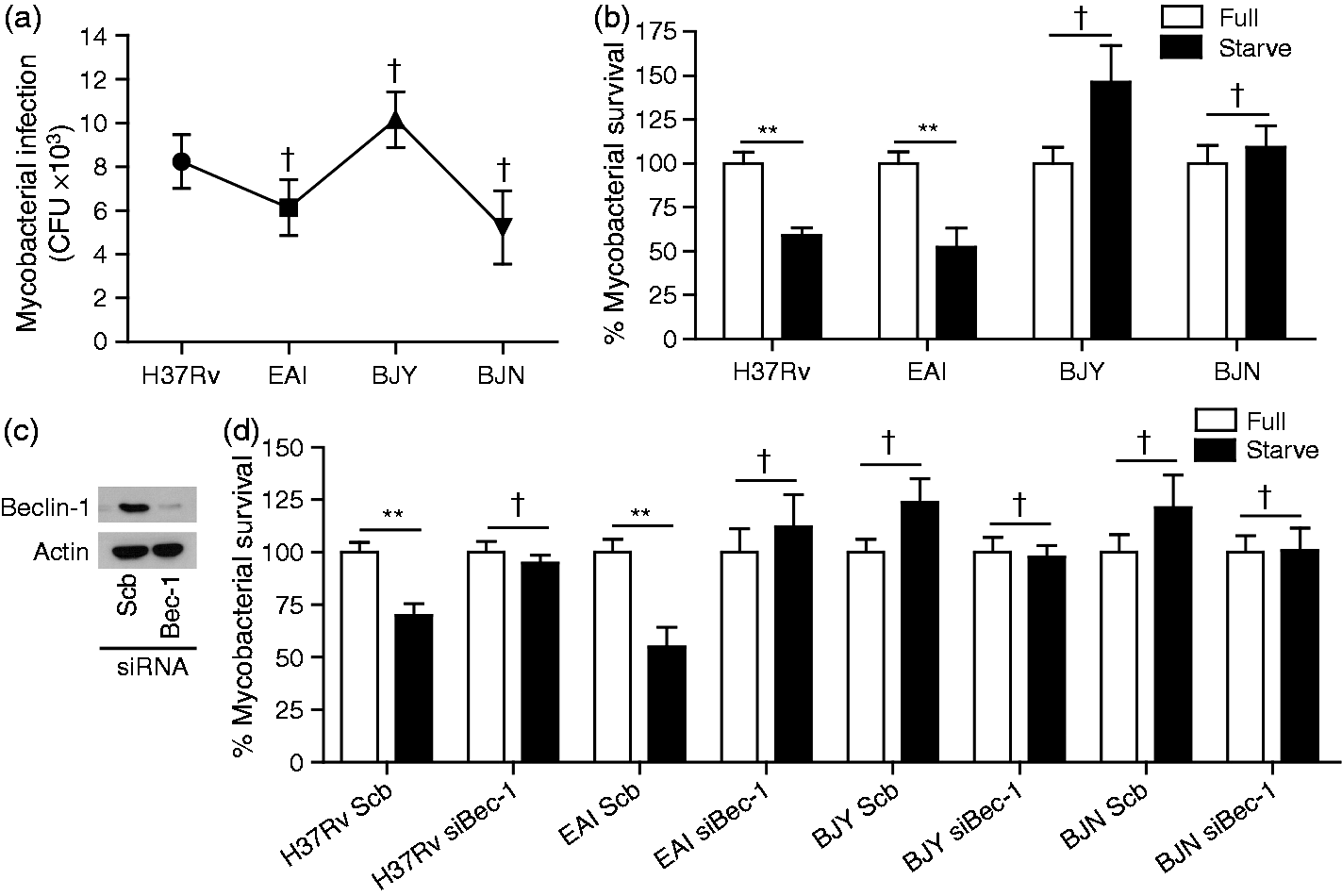
The Beijing strains avoid autophagic elimination not by inhibiting autophagy-mediated acidification of their phagosomes
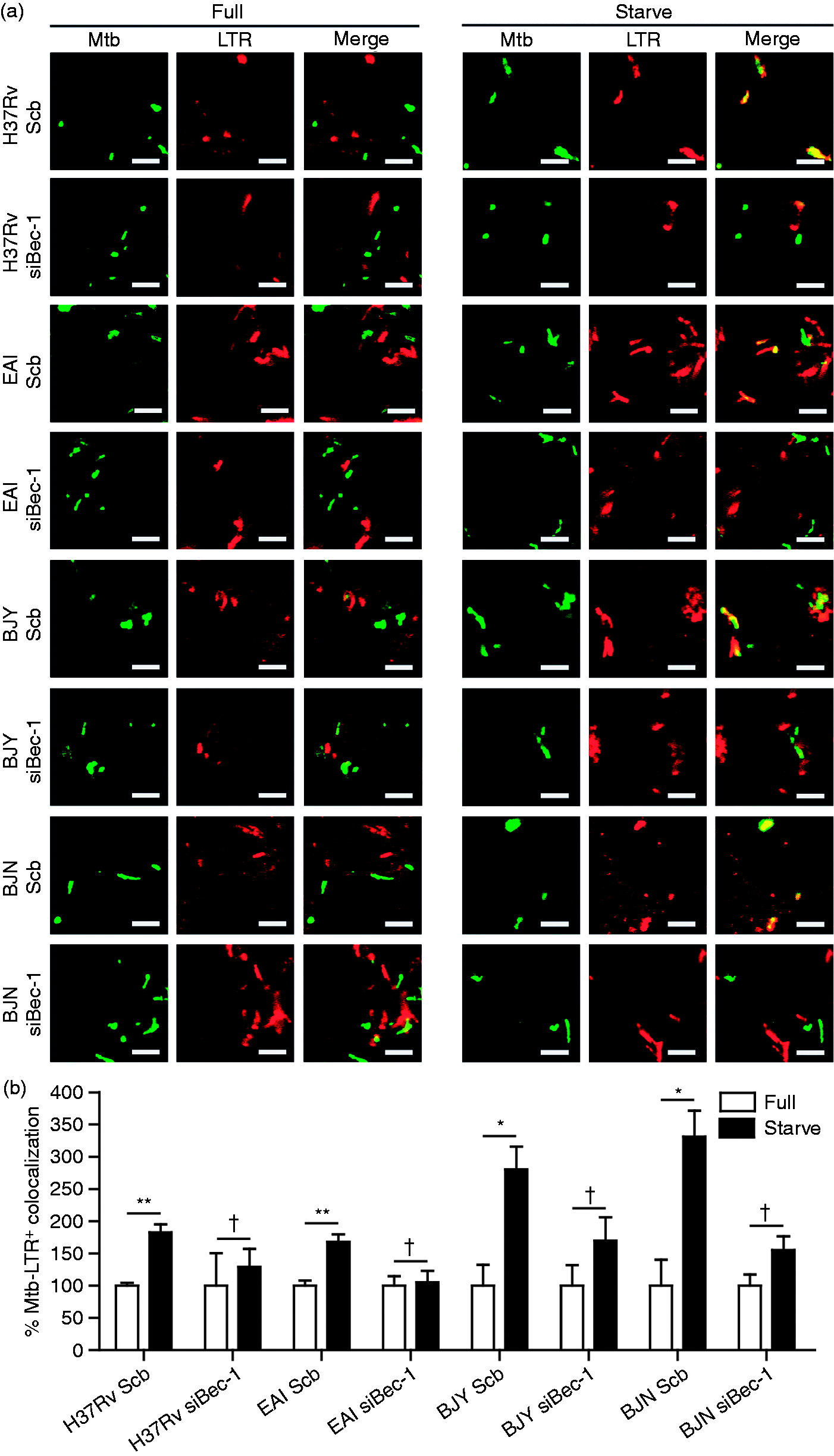
As our results showed the greater ability of the Beijing strains to resist starvation-induced autophagic killing by host cells (Figure 1), we set out to determine the molecular mechanism involved. We first examined whether the ability of the Beijing strains to evade autophagic restriction is due to their ability to inhibit autophagy-mediated acidification of their phagosomes. To determine this, we performed confocal microscopy analysis for colocalization of mycobacteria with LTR dye. The results showed that acidification of M. tuberculosis H37Rv phagosomes is enhanced upon autophagy induction by starvation, as previously reported for reference strains (Figure 2A, B).14,15,37,46 In addition, the induction of autophagy by starvation increases the phagosomal acidification of the EAI strain, albeit not statistically significantly (Figure 2A, B). Surprisingly, the acidification of the Beijing strains (BJY and BJN) also appeared to be substantially enhanced upon autophagy induction by starvation (Figure 2A, B). Thus, these results indicated that even though the Beijing strains are able to inhibit the autophagic control by host cells, these bacteria do so not by simply inhibiting the autophagy-mediated acidification of their phagosomes but through another mechanism.
The Beijing strains inhibit starvation-induced autophagic killing not by blocking autophagy-mediated acidification of their phagosomes. (A, B) RAW264.7 macrophages were infected with Alexa-488-labeled mycobacteria at an MOI of 10 for 15 min and subsequently chased for 1 h in complete media. LTR dye was used to stain acidic compartments. Cells were then induced to undergo autophagy by starvation for 2 h. Cells were then processed for confocal microscopy analysis for the colocalization of mycobacteria with LTR. Data are mean ± SEM from at least three independent experiments. At least 50 phagosomes per condition per independent experiment were quantified. **P < 0.01, *P < 0.05, †P ≥ 0.05, all relative to the full control set to 100%. EAI, the East African Indian strain with intact pks15/1; BJY, the Beijing strain with intact pks15/1; BJN, the Beijing strain without intact pks15/1.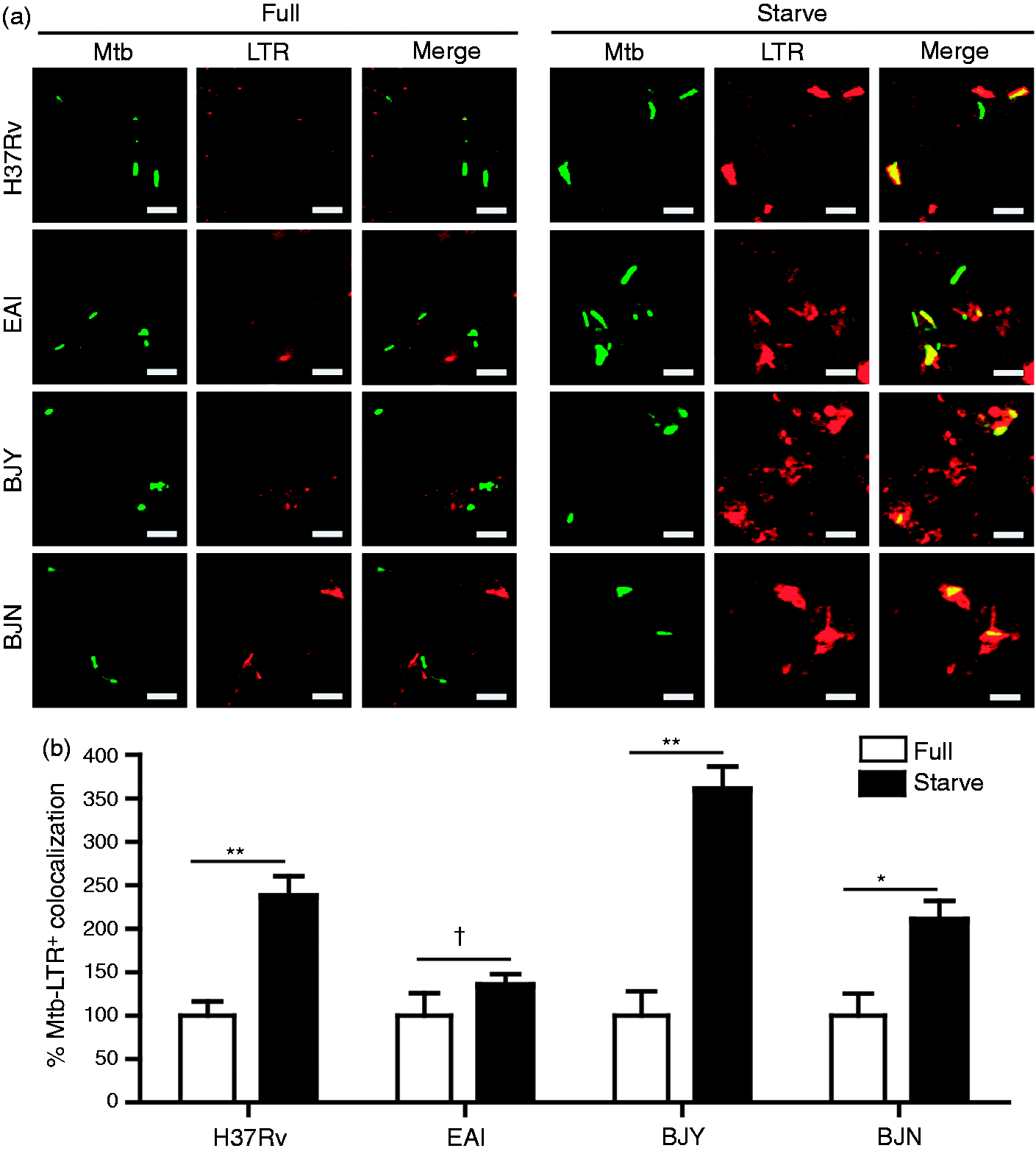
The ability of the Beijing strains to escape autophagic restriction is not conferred by their capacity to block host autophagic flux
In addition to the direct killing mechanism mediated by autophagy whereby mycobacteria are captured into autophagosomes and then delivered into acidic lysosomes, autophagy can eliminate mycobacteria using another indirect mechanism whereby the innocuous cytosolic precursors are captured into autophagosomes, which, during the process of maturation into autolysosomes, partially digest these precursors into killing peptides that are subsequently delivered to mycobacteria via fusion of the autophagosomes/autolysosomes with M. tuberculosis phagosomes.
15
Thus, the Beijing strains may evade autophagic killing through their differential ability to block the maturation of other population of cytosolic autophagosomes not containing mycobacteria. To test this possibility, autophagic flux in host cells was measured by fluorescent microscopy analysis for the conversion of RFP+GFP+–LC3 puncta (autophagosomes) to RFP+GFP−–LC3 puncta (autolysosomes). The results showed that the maturation of autophagosomes into autolysosomes is similarly inhibited in host macrophages infected with different M. tuberculosis strains when compared with that of the uninfected cells (Figure 3A, B, white bar) as demonstrated by an increase in the number of RFP+GFP+–LC3 puncta in conjunction with no increase in the number of RFP+GFP−–LC3 puncta. The block in autophagic flux upon infection was further demonstrated when cells were subjected to autophagy induction by starvation (Figure 3A, B, black bar). These results indicated that all of the tested strains and the reference M. tuberculosis H37Rv possess comparable ability to retard autophagic flux in host macrophages. Thus, the greater ability of the Beijing strains to resist autophagic elimination cannot be attributed to their ability to inhibit autophagic maturation in host cells.
Evasion of starvation-induced autophagic elimination by the Beijing strains is not a result of their differential ability to block autophagic flux in host cells. (A, B) RAW264.7 cells were transfected with cDNAs encoding RFP–GFP–LC3. Transfected cells were then infected with Alexa-405-labeled mycobacteria for 15 min and subsequently chased for 1 h in complete media. Cells were induced to undergo autophagy by starvation for 2 h. Cells were then processed for confocal microscopy (Z-Stacks) analysis for the number of LC3 puncta per cell. Data are mean ± SEM combined from two independent experiments. The number of RFP+GFP+–LC3 (autophagosomes) and RFP+GFP−–LC3 (autolysosomes) were quantified in Z-Stacks images of 30 cells per condition per independent experiment. **P < 0.01 and †P ≥ 0.05, all relative to full and starvation of uninfected control. EAI, the East African Indian strain with intact pks15/1; BJY, the Beijing strain with intact pks15/1; BJN, the Beijing strain without intact pks15/1.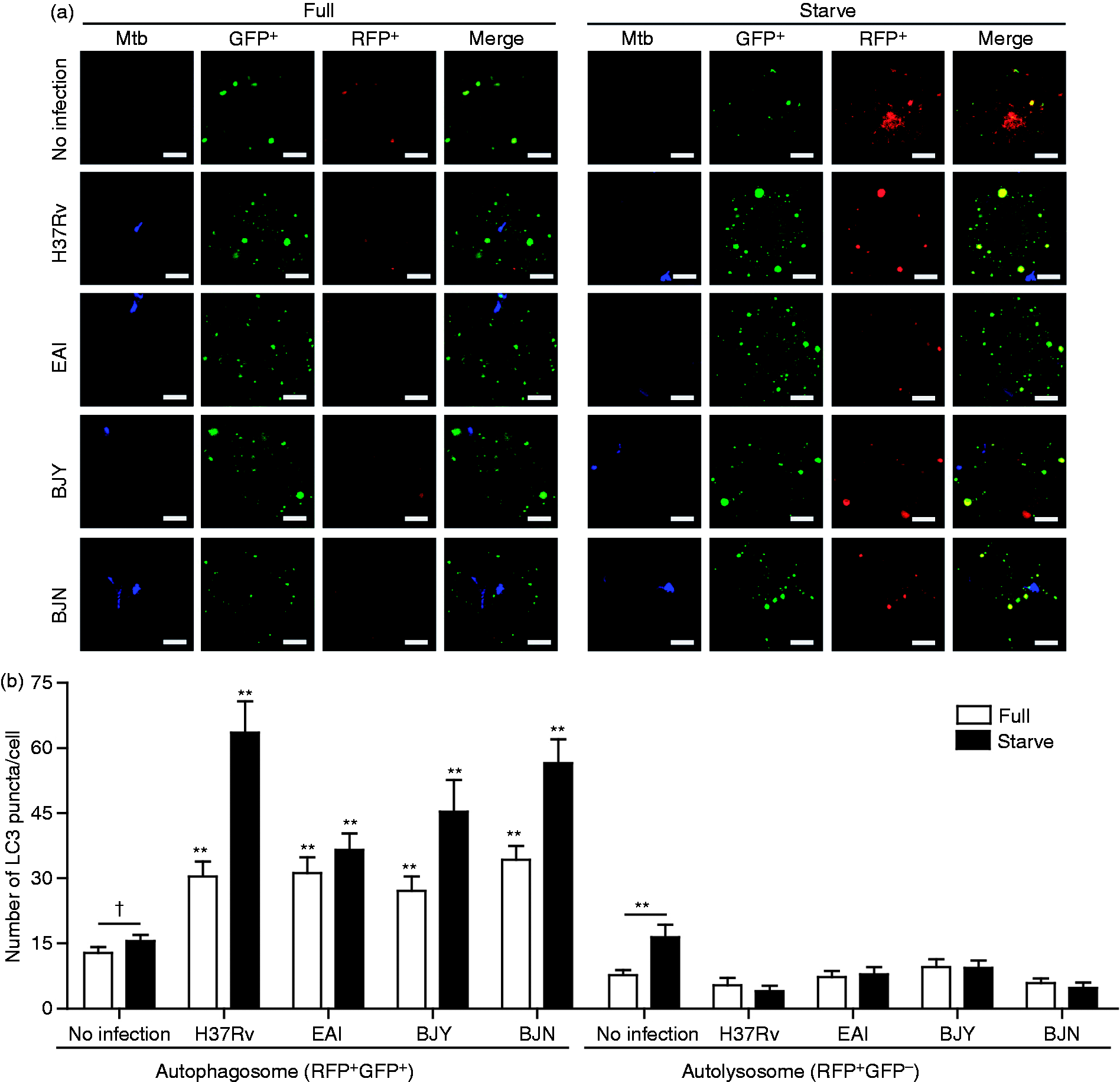
The Beijing strains evade starvation-induced autophagic control by suppressing autophagolysosome biogenesis
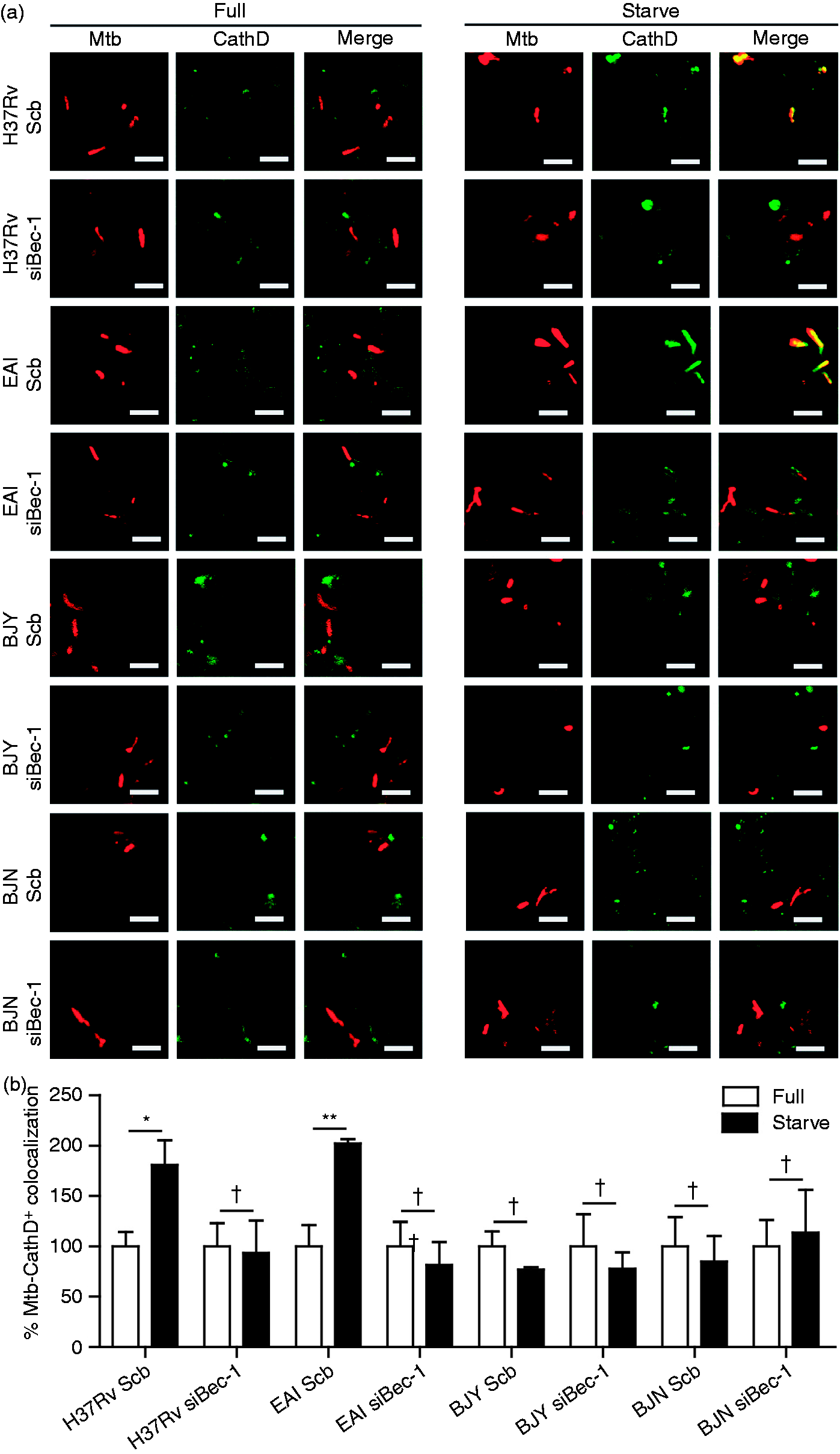
Some bacteria such as Helicobacter pylori and Mycobacterium marinum were shown to reside in compartments that are acidified but lack a lysosomal marker cathepsin D in response to autophagy,48–50 indicating a defect in autophagolysosome biogenesis and thus sparing these bacteria from elimination. Therefore, we sought out to determine whether the Beijing strains may possess a similar ability. As a result, we analyzed the colocalization of different strains of M. tuberculosis with LTR and cathepsin D upon autophagy induction by starvation. Confirming our results shown in Figure 2, acidification of phagosomes containing different strains of M. tuberculosis are substantially augmented (LTR+) upon treatment of cells with starvation (Figure 4A, C). However, while there was enhanced colocalization of the reference strain H37Rv and the EAI strain with cathepsin D (CathD+) in response to autophagy induction by starvation (Figure 4B, D), there was no increase in the colocalization of the Beijing strains (BJY and BJN) with cathepsin D (CathD+) observed upon starvation treatment (Figure 4B, D). These data indicated that even though their phagosomes are efficiently acidified, the Beijing strains possess an ability to suppress autophagolysosome biogenesis in response to autophagy induction. Note that the anti-cathepsin D Ab used can recognize both the immature and mature forms of cathepsin D and hence the lack of increased colocalization of the Beijing phagosomes with cathepsin D may include both the mature and immature forms. To further confirm autophagy involvement in this process, we depleted Beclin-1 in RAW264.7 macrophages and assessed the colocalization of different mycobacteria with LTR and cathepsin D. Our results showed that autophagy is required for the increased phagosome acidification of all M. tuberculosis strains (LTR+) upon starvation (Figure 5A, B). However, while there was an increase in the colocalization of M. tuberculosis reference strain H37Rv and the EAI strain with cathepsin D (CathD+) upon starvation treatment and that this process depends on autophagy (Figure 6A, B), there was no increase in CathD+ colocalization with the Beijing strains upon starvation treatment, even in autophagy efficient cells (Figure 6A, B). Altogether, these data indicate that the Beijing strains possess a greater ability to evade starvation-induced autophagic killing by their higher capacity to inhibit autophagolysosome biogenesis as demonstrated by their suppression of the enhanced acquisition of cathepsin D into their phagosomes upon autophagy induction in host cells.
The Beijing strains escape from starvation-induced autophagic elimination by suppressing autophagolysosome biogenesis. (A, C) RAW264.7 cells were infected with Alexa 405-labeled mycobacteria (pseudocolored green) for 15 min and subsequently chased for 1 h in complete media. Acidic compartments were stained with LTR. Cells were then induced to undergo autophagy by starvation for 2 h. Cells were then fixed and processed for confocal microscopy analysis. (B, D) RAW264.7 cells were infected with Alex 405-labeled mycobacteria (pseudocolored red) and processed as described above. Cells were then fixed and stained for cathepsin D. Data are mean ± SEM from at least three independent experiments. At least 50 phagosomes per condition per independent experiment were quantified. **P < 0.01, *P < 0.05, †P ≥ 0.05, all relative to the full control set to 100%. EAI, the East African Indian strain with intact pks15/1; BJY, the Beijing strain with intact pks15/1; BJN, the Beijing strain without intact pks15/1. Autophagy is required for the starvation-enhanced acidification of the M. tuberculosis reference strain H37Rv and the EAI strain phagosomes. (A, B) RAW264.7 cells were transfected with siRNAs against Beclin-1 or scramble (Scb) control. After 48 h of transfection, cells were infected with Alexa 405-labeled mycobacteria (pseudocolored green) and processed as in Figure 4 (A, C). Data are means ± SEM from at least three independent experiments. At least 50 phagosomes per condition per independent experiment were quantified. **P < 0.01, *P < 0.05, and †P ≥ 0.05, all relative to the full control set to 100%. EAI, the East African Indian strain with intact pks15/1; BJY, the Beijing strain with intact pks15/1; BJN, the Beijing strain without intact pks15/1. The increased cathepsin D acquisition in response to starvation observed with the M. tuberculosis reference strain H37Rv and the EAI strain depends on autophagy. (A, B) RAW264.7 cells were transfected with siRNAs against Beclin-1 or scramble (Scb). After 48 h of transfection, cells were then infected with Alexa 405-labeled mycobacteria (pseudocolored red) and processed for cathepsin D staining as in Figure 4 (B, D). Data are means ± SEM from at least three independent experiments. At least 50 phagosomes per condition per independent experiment were quantified. **P < 0.01, *P < 0.05, and †P ≥ 0.05, all relative to the full control set to 100%. EAI, the East African Indian strain with intact pks15/1; BJY, the Beijing strain with intact pks15/1; BJN, the Beijing strain without intact pks15/1.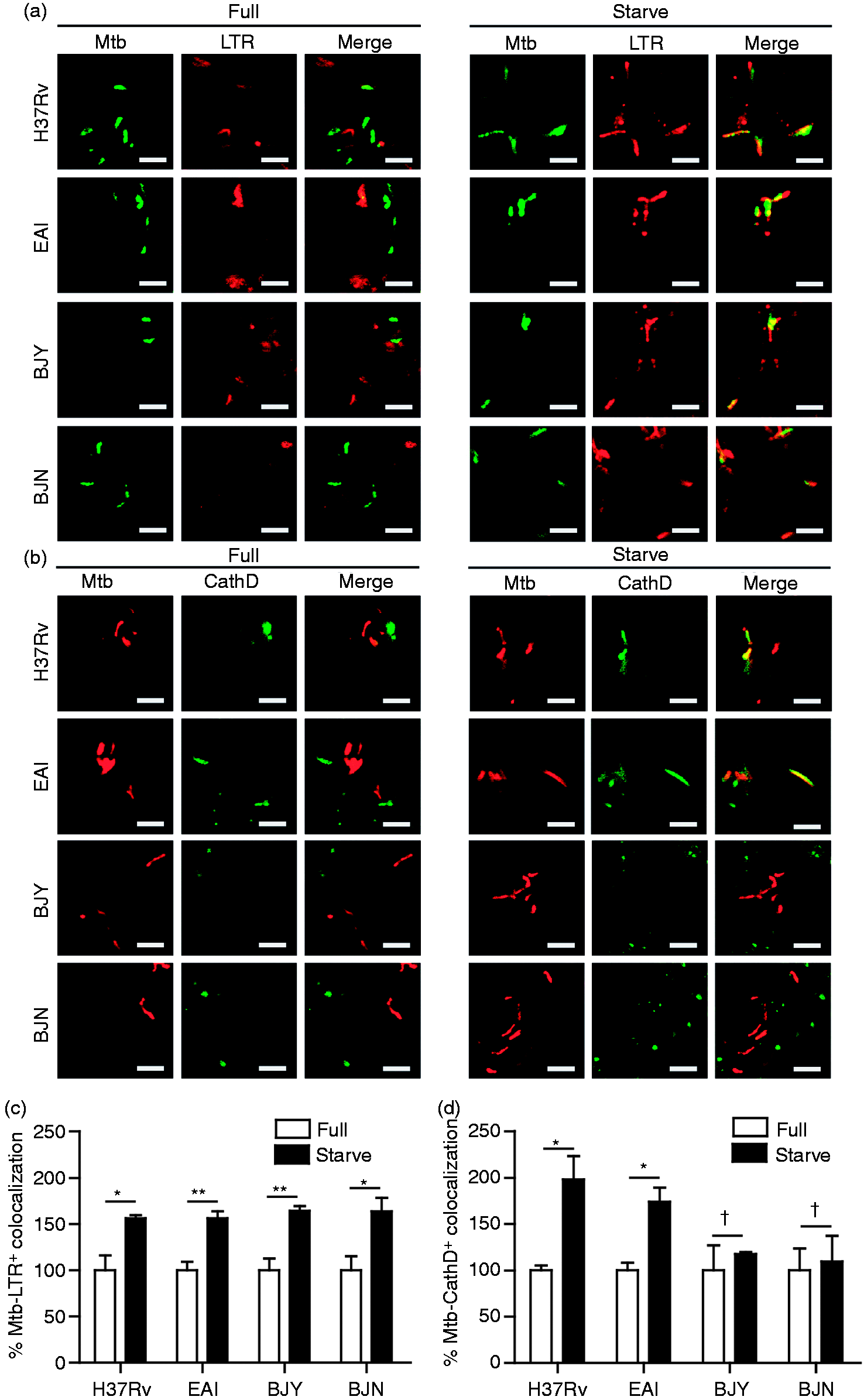
Taken together, our results show that there is a variation in the capacity of different M. tuberculosis strains to resist starvation-induced autophagic killing in host macrophages, of which the Beijing strains possess a unique capability to evade autophagic elimination upon autophagy induction. The Beijing strains do so not by simply inhibiting the autophagy-mediated acidification of their phagosomes or by blocking the autophagic flux in host cells, but by inhibiting autophagolysosome biogenesis, as demonstrated by their ability to suppress the increased acquisition of cathepsin D into their compartments upon autophagy induction by starvation and thus escape from autophagic restriction.
Discussion
Autophagy has been demonstrated as an important innate immune defense mechanism utilized by host cells for the elimination of various intracellular bacteria, parasites and viruses.1,9–11 In addition, induction of autophagy by starvation or other autophagy inducers has been extensively shown to result in the killing of M. tuberculosis by disabling the block in phagolysosomal biogenesis imposed by the tubercle bacilli.12–17 As the ability of autophagy in the restriction of M. tuberculosis has been observed in studies using laboratory reference strains but has not been characterized in detail in cells infected with the clinical strains, especially those belonging to the Beijing family, which has previously been shown to be hyper-virulent (associated with their higher ability to survive in host macrophages and cause higher bacterial load and mortality in animal models),31–36 we set out to examine the autophagic killing capacity of host cells against different strains of M. tuberculosis. In our present study, we uncovered the previously unrecognized ability of M. tuberculosis Beijing strains to evade starvation-induced autophagic elimination. In addition, we have determined a possible underlying mechanism in which the Beijing strains can escape starvation-induced autophagic killing by inhibiting autophagolysosome biogenesis, as demonstrated by their ability to suppress the enhanced acquisition of cathepsin D into their compartments. As autophagy was previously shown to be a key determinant of host resistance against mycobacterial infection,20,21,23–28 our findings may explain the hyper-virulent phenotypes associated with the Beijing family,31–36 which are thought to have resulted from their higher ability to survive in host macrophages, but the underlying mechanism was previously unknown.
Consistent with previous findings that showed the effect of starvation-induced autophagy in the elimination of M. tuberculosis reference strains,12–17 we also observed that starvation treatment of infected RAW macrophages results in the restriction of the laboratory reference strain M. tuberculosis H37Rv. In contrast to that of the reference strains, we observed a previously unrecognized ability of M. tuberculosis Beijing strains to avoid autophagic killing by blocking autophagolysosome biogenesis. The ability to interfere with the autophagy pathway of host macrophages possessed by the Beijing strains is similar to those possessed by and previously reported for H. pylori and M. marinum.48–50 These bacteria were also found to escape from autophagic elimination by inhibiting autophagolysosome biogenesis in host cells in response to autophagy induction, as determined by a decrease in enhanced cathepsin D acquisition into their compartments. Cathepsin D, an aspartic endopeptidase, is synthesized in the rough endoplasmic reticulum as preprocathepsin D and then targeted to various compartments such as phagosomes, autophagosomes, endosomes and lysosomes as procathepsin D.51–53 Upon transport, inactive procathepsin D is converted into active form of cathepsin D by acidic pH and thus the presence of cathepsin D precursors (and other lysosomal acid hydrolases) in acidified compartments is necessary for their degradative function. Our results showed that although the acidification of M. tuberculosis Beijing phagosomes are efficiently increased upon autophagy induction by starvation, these was no increase in the acquisition of cathepsin D into their phagosomes upon starvation treatment, indicating a defect in autophagolysosome biogenesis.
The mechanism by which the Beijing strains inhibit autophagolysosome biogenesis is under investigation by our laboratory. Among several mycobacterial factors previously shown to be involved in M. tuberculosis survival in host cells, phenolic glycolipid (PGL) synthesized by a polyketide synthase encoded by intact pks15/1 was demonstrated to reduce the production of Th1-type cytokines,47,54 some of which have been shown to induce autophagy. 1 However, our data showed no significant relationship between the presence of intact pks15/1 and the ability of mycobacteria to resist starvation-induced autophagic elimination, thus indicating that PGL is not the factor that renders the Beijing strains to escape autophagic restriction. Interestingly, in the case of H. pylori, which was also shown to evade autophagic killing by residing in acidic compartments that lack cathepsin D in response to autophagy,48–50 it was previously reported that the maturation of procathepsin D into the mature form, as well as its sorting in mammalian cells, can be impaired by H. pylori vacuolating toxin. 55 Whether M. tuberculosis Beijing strains possess a similar or equivalent protein is not known. As a recent whole-genome sequencing of strains belonging to the M. tuberculosis Beijing family has been conducted, 56 mining and comparing sequencing data between Beijing and non-Beijing strains may give insight into potential candidate genes that may play a role in the autophagic evasion. In addition to the bacterial factor, two host proteins, Rab8b and its downstream effector TANK-binding kinase 1 (TBK1), have been shown to play prominent roles in autophagy-mediated mycobacterial phagosome maturation, as demonstrated by decreased mycobacterial phagosome acidification and cathepsin D acquisition upon siRNA-mediated depletion of Rab8b or pharmacological inhibition of TBK1. 37 Whether the Beijing strains are able to interfere with Rab8b and TBK1 functions in autophagy-mediated mycobacterial phagosome maturation is a subject of our study.
Besides our principle finding, discussed above, our data also showed that the acquisition of cathepsin D into the EAI phagosomes is substantially enhanced upon autophagy induction, similar to what has previously been observed with M. tuberculosis reference strains.12–17 Interestingly, previous studies showed that the EAI strains possess a higher ability to stimulate TNF-α synthesis in infected host cells when compared with that of the Beijing strains.40,57 As TNF-α has been shown to be a potent autophagic inducer, 1 the enhanced production of TNF-α upon EAI strain infection may augment the autophagy induction level in our system and may thus help increase the acquisition of cathepsin D into the EAI phagosomes. Whether the EAI strain possesses greater ability to induce host autophagy as a result of increased TNF-α production awaits further examination. If the EAI strain is found to enhance host autophagy, this may, in turn, explain the low virulence phenotypes associated with this M. tuberculosis family, as demonstrated by their reduced transmissibility, low level of growth inside macrophages, and decreased bacterial burden (100-fold lower when compared with that of the Beijing strains) and mortality in mouse models.29,38 In addition, we also observed an enhanced growth of the Beijing strains inside host cells upon autophagy induction, albeit not a statistically significant increase, and this effect was decreased in cells deficient of Beclin-1 (Figure 1B, D). Whether the Beijing strains can, in addition to resisting autophagic killing by host cells, subvert autophagy pathway for their own growth inside macrophages await further investigation.
Footnotes
Funding
This work was supported by Thailand Research Fund, Office of the Higher Education Commission, and Mahidol University; National Science and Technology Development Agency; and Faculty of Science, Mahidol University.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.