
Abstract
The acquisition of innate immune response is requisite to having bona fide differentiation of airway epithelium. Procedures developed to differentiate lung airway from human pluripotent stem cells (hPSCs) have demonstrated anecdotal evidence for innate immune response, but an in-depth exploration of response levels is lacking. Herein, using an established method of airway epithelial generation from hPSCs, we show that hPSC-derived epithelial cells are able to up-regulate expression of TNFα, IL8 and IL1β in response to challenge with bacterial endotoxin LPS, but lack response from genes associated with innate immune response in other cell types. Further, stimulation of cells with TNF-α resulted in auto-induction of TNFα transcript, as well as cytokine responses of IL8 and IL1β. The demonstration of innate immune induction in hPSC-derived airway epithelia gives further strength to the functionality of in vitro protocols aimed at generating differentiated airway cells that can potentially be used in a translational setting. Finally, we propose that innate immune challenge of airway epithelium from human pluripotent stem cell sources be used as a robust validation of functional in vitro differentiation.
Keywords
Introduction
The innate immune system is the body’s first line of defense against infection. In human lungs, epithelial cells mediate direct contact between the environment and the large surface area of the airways. These cells can respond to pathogens inhaled during respiration by secreting cytokines and chemokines, thus regulating inflammation. Cytokines produced and released by airway epithelial cells during infection include IL-1α and IL-1β, and IL-8 and TNF-α. 1
Recently, expanding upon techniques established to differentiate airway epithelial cells from human embryonic stem cells (hESCs) and induced pluripotent stem cells (hiPSCs), collectively termed hPSCs for human pluripotent stem cells, 2 it was demonstrated that functional airway epithelium can be generated by extending culture times and enriching for differentiated epithelia using a two-step air–liquid interface procedure. 3 This study showed that hPSC-derived lung epithelia express airway markers and become ciliated upon culture in air–liquid interface systems. Thus far, there has only been anecdotal evidence to support the ability of in vitro-induced airway epithelial cells to mount an innate immune response to bacterial endotoxin. 3 In this study, we have used an established method of directed differentiation of hPSCs to lung epithelial cells to obtain functional airway cells, and demonstrate their innate immune function by assessing gene and protein expression of mediators of the innate immune system in response to challenge with the bacterial endotoxin, LPS and stimulation with the pro-inflammatory cytokine TNF-α. We demonstrate that hPSC-derived lung epithelia can mount an innate immune response by increasing expression of TNF-α transcript and protein, and by augmenting transcript levels of IL1β and IL8 in response to challenge of the innate immune system, with confirmation by Affymetrix (Santa Clara, CA, USA) expression array profiling. These findings form the basis of creating a robust validation platform for airway cells differentiated from hPSCs and provide the first evidence of innate immune response from cells differentiated from pluripotent sources.
Materials and methods
Human embryonic stem cell culture and differentiation
Two human embryonic stem (ES) cell lines, H9 and CA2, 4 were maintained in mTeSR1 (Stem Cell Technologies, Vancouver, BC, Canada) on Matrigel (BD Biosciences, San Diego, CA, USA) and passaged 1:2 by dissociation with Collagenase IV (Sigma Aldrich, Oakville, ON, Canada) every 5–7 d. Differentiation was performed as previously described using Matrigel-coated Costar 3470 (Corning, Tewksbury, MA, USA) inserts in which air–liquid culture was executed. 3 Cells were split once in air–liquid [secondary air–liquid interface culture (2’ALI)] as described, 3 and stimulated for 2, 5 or 18 h with 5 µg/ml cell culture-grade bacterial LPS from Escherichia coli 0111:B4 (Sigma, St. Louis, MO, USA) or 10 ng/ml recombinant human TNF-α (Sigma).
RNA extraction, cDNA synthesis and quantitative PCR
RNA was extracted using the Norgen Total RNA Extraction and Purification Kit according to the manufacturer’s protocol (Norgen Biotek, Thorald, ON, Canada). cDNA was synthesized from 1 µg RNA per sample with an optimized blend of random and oligo(dT) primers using the qScript cDNA Synthesis Kit (Quanta BioSciences, Gaithersburg, MD, USA). Quantitative RT-PCR (qPCR) was performed using the GoTaq qPCR master mix (Promega, Madison, WI, USA) with fluorescence read in the SYBR Green (FAM) channel on a Bio-Rad CFX-96 thermocycler (Bio-Rad, Hercules, CA, USA) running a one-step extension and annealing protocol at 62℃ for 20 s following denaturation for 15 s at 94℃ for 40 cycles. A melt curve analysis was run for each reaction from 65℃ to 94℃ to verify product specificity (Figure 1).
Experimental layout and quality control. (A) hPSCs were maintained in chemically defined media and differentiated to airway epithelial cells using a protocol involving air–liquid interface culture, where they acquire an epithelial phenotype as documented by ZO-1 confocal microscopy. Following differentiation, cells were challenged with LPS or TNF-α and expression from candidate response genes was evaluated. Initial semiquantitative PCR for TNFα and IL8 showed increased expression following challenge with LPS. Images are taken from actual experiments but are not directly comparable. (B) Primer sequences and quality control of amplicons (melt curve analysis) evaluated in this study show single transcript amplification without evidence of primer dimers. Melt curve is measured by increasing temperature (x-axis) over time and measuring a steep decrease in fluorescence signal (y-axis) around the specific melting temperature of each PCR product.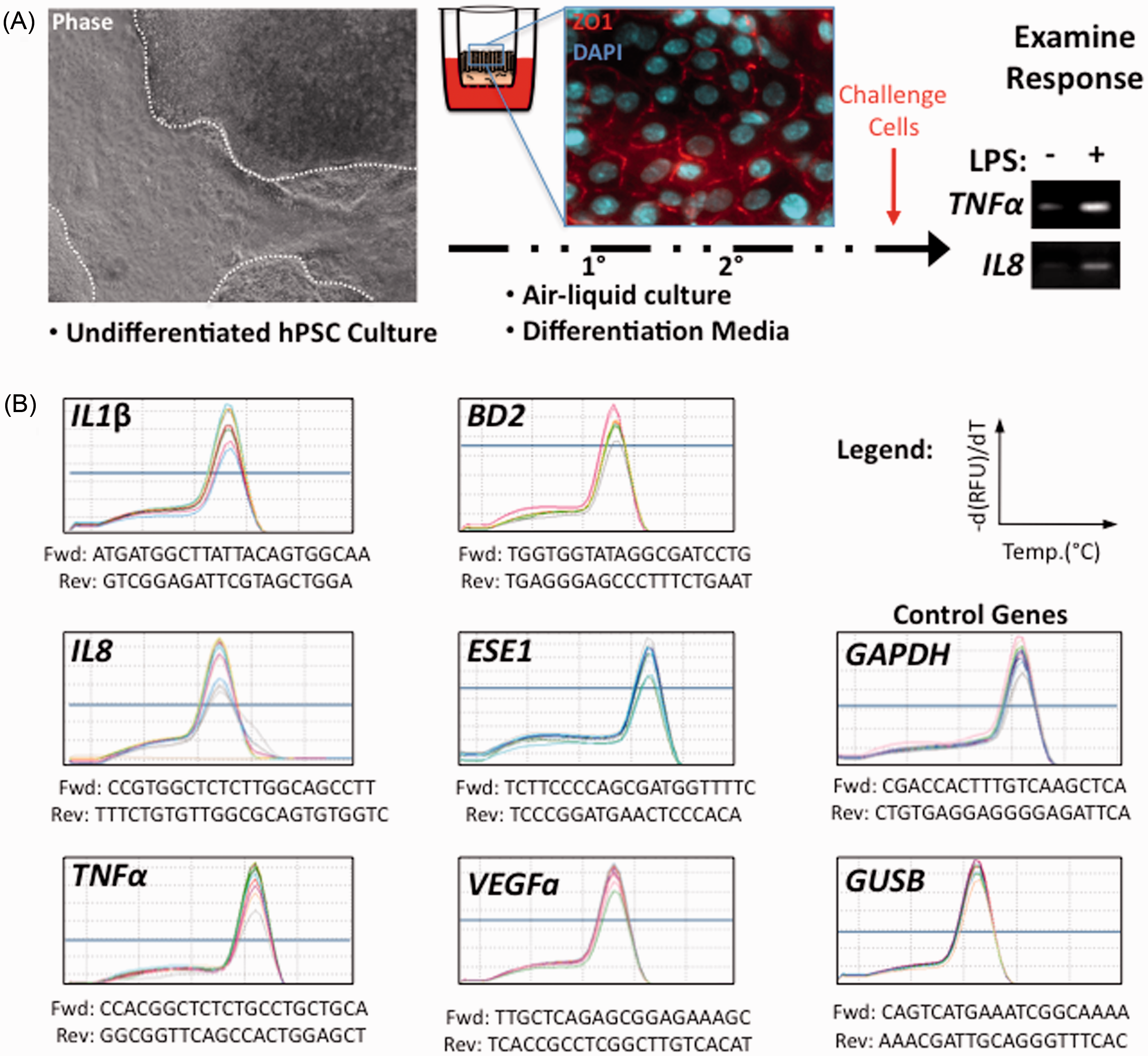
Expression analysis by qPCR
Expression levels were compared with the control genes GAPDH and GUSB using the comparative Ct method (2−ΔΔCt). GAPDH and GUSB levels were stable with respect to each other and with respect to a third endogenous control gene, TBP. However, a fourth control, ribosomal 18S rRNA (18S) was down-regulated following LPS challenge and was thus excluded as a control from this study (please refer to the ‘Results’ for further details). Statistical significance was determined by examining expression relative to untreated controls using a two-tailed Student’s t-test. All samples represent a minimum of three replicates and graphs show mean values ± SEM.
ELISA
Quantification of TNF-α from conditioned media collected from 100-µl washes of the apical supernatant in growth media was performed using Human TNFα ELISA MAX sets following the manufacturer’s instructions (BioLegend, San Diego, CA, USA). Samples were diluted 1:5 in Assay Diluent A (BioLegend). Similarly, TNF-α standards were reconstituted in Assay Diluent A and further diluted 1:5 in Assay Diluent A for experimental analysis, determined empirically to be the most accurate method of quantification. TNF-α levels in unknowns were calculated based on the generated equation from the standard curve.
Confocal microscopy
ALI cultures were stained en face following acetone fixation with anti-ZO-1 (TJP1) Ab (HPA001636; Sigma) at 1:200 and counterstained with DAPI for nuclear visualization. Images were captured on an Olympus spinning disk confocal microscope (Olympus, Tokyo, Japan) running Metamorph software (Molecular Devices, Sunnyvale, CA, USA), overlays were made using the open source ImageJ software suite (NIH, Bethesda, MD, USA).
Microarray expression profiling
Affymetrix array data using the Human Gene 1.0 ST Array platform (Affymetrix, Santa Clara, CA, USA) were normalized using the Robust Multichip Averaging algorithm and Log2 transformed using Partek Genomics Suite 6.6 software (Partek, St. Louis, MO, USA). Normalized arrays were hierarchically clustered based on a specified gene list. In cases where multiple probe sets existed for one gene, expression values were averaged. This dataset has been uploaded to Gene Expression Omnibus (GEO) under the accession number GSE59472, to allow access and data mining for the broader scientific community.
Results
hPSC differentiation to airway epithelium results in quantitative innate immune responses to LPS challenge
Lung epithelial differentiation was carried out using hPSCs maintained in chemically defined mTeSR1 primed with a 5-d treatment of activin-A (R&D Systems, Minneapolis, MN, USA) at 100 ng/ml and differentiated on transwell inserts using a dual air–liquid interface system in 80% knockout DMEM supplemented with 20% knockout serum replacement (Life Technologies, Burlington, ON, Canada) containing non-essential amino acids and
Gene expression was initially evaluated in hPSC-derived airway cells at both 2 and 5 h following stimulation by qPCR. Both LPS and TNF-α elicited robust increases in the expression of both IL1β and IL8 at 5 h (Figure 2A, B). LPS had a more modest effect in increasing TNFα transcript levels, whereas this gene underwent robust auto-stimulation in response to TNF-α challenge at both time points measured (Figure 2B). At 2 h no increases were observed following LPS challenge (Figure 2A), but transcription of IL1β, IL8 and TNFα were all up-regulated in response to TNF-α at this early time point (Figure 2B). Interestingly, expression of ESE1, which is described as being up-regulated by TNF-α in bronchial epithelial cells,
8
was not augmented in hPSC-derived airway epithelium (Figure 2B). Similarly, expression of VEGFA, which has been documented to increase in response to TNF-α in primary human nasal cells and in the serum of mice challenged systemically with bacterial endotoxin,9,10 was not observed (Figure 2B). This was also the case for BD2 (Figure 2), which has been shown to be up-regulated in response to TNF-α and IL-1β stimulation but not in response to IL6, in cultured primary human airway epithelial cells.
11
Gene expression changes following challenge with LPS and TNF-α at 2 and 5 h. (A) Increases in expression were observed for IL1β, IL8 and TNFα following LPS stimulation at 5 h only. (B) Expression increases were observed for IL1β, IL8 and TNFα following stimulation with TNF-α at both 2 and 5 h. Expression is plotted as fold increase over carrier (PBS) treated controls set to 1 (gray dashed line). *p<0.05, **p<0.01, ***p<0.001.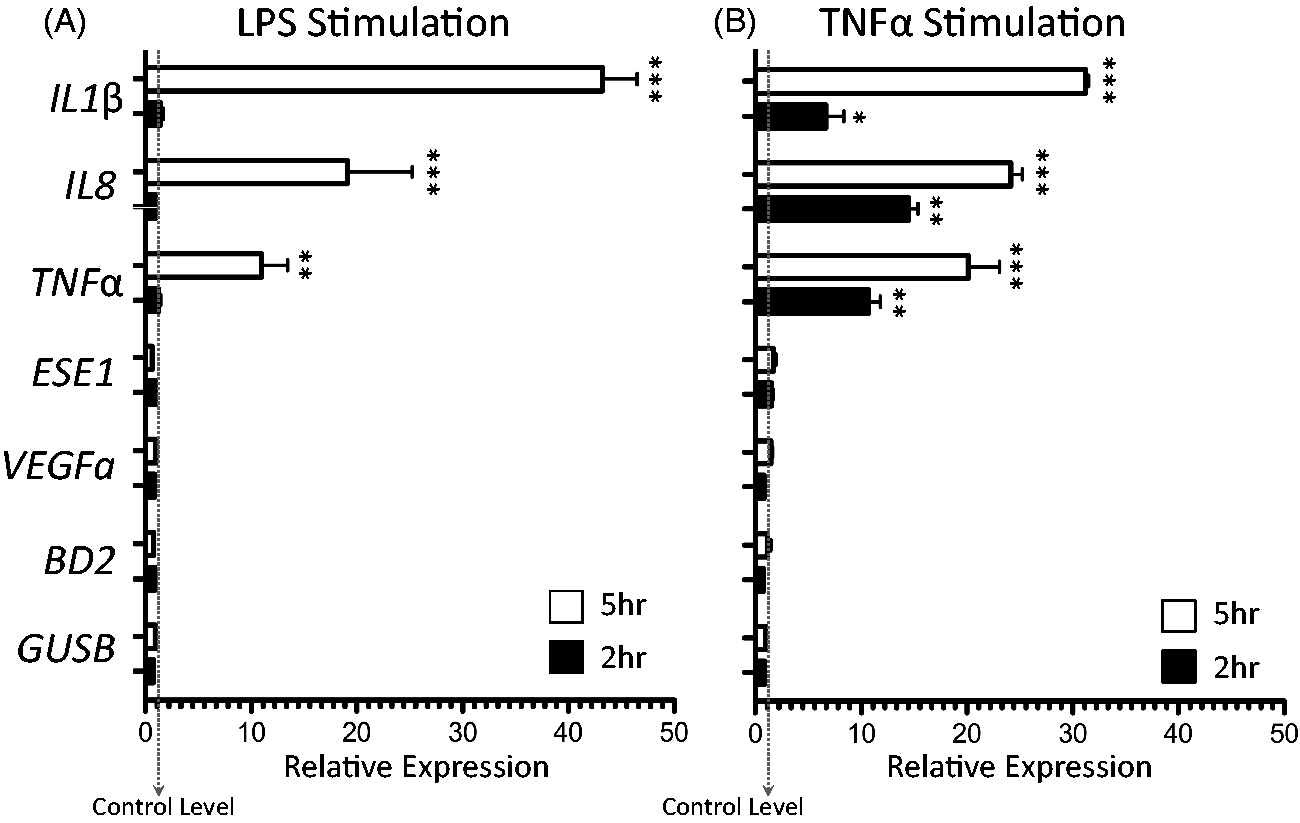
In addition to genes known to be involved in the innate immune response of the airway, we examined the expression of IL6, which has been described to be up-regulated in macrophages and T cells in response to LPS and TNF-α in macrophages and T-cells.12,13 Informatively, IL6 transcript was not observed to be up-regulated in our system. The lack of response of IL6 indicates that our differentiation protocol is not producing either of these cell types and innate response is most likely being mounted by airway epithelial cells. Numerous additional genes expressed by airway epithelia such as SP-A and SP-D, which also have a role in innate immunity, 14 showed no augmentation in expression in response to LPS or TNF-α. The full battery of genes assayed, but not modulated by either LPS or TNF-α include IL6, SP-A, SP-D, IFNG, iNOS, TGF-β, MUC5B and CC10/CC16.
In order to validate our findings by qPCR we subjected six samples, two controls and two samples stimulated with either LPS or TNF-α, respectively, to Affymetrix Gene Expression Array profiling (GEO accession: GSE59472). Analysis of expression was able to verify augmented IL1β, IL8 and TNFα expression, but also demonstrated a robust increase in a curated gene list of innate immune markers of the airway: CCL4, CCL5, CCL8, CCL12, CX3CL1, CXCL11, CXCR4, ICAM1, VCAM1, IL32 and IL1α (Figure 3).
Heat map demonstrating expression changes in candidate genes chosen to reflect airway epithelial responses to innate immune challenge. Note the robust up-regulation in all markers examined versus control airway differentiated ES cells. Hierarchical clustering tightly groups TNF-α and LPS challenged cells away from non-challenged controls. Scale bar shows color coded log2 changes in expression level.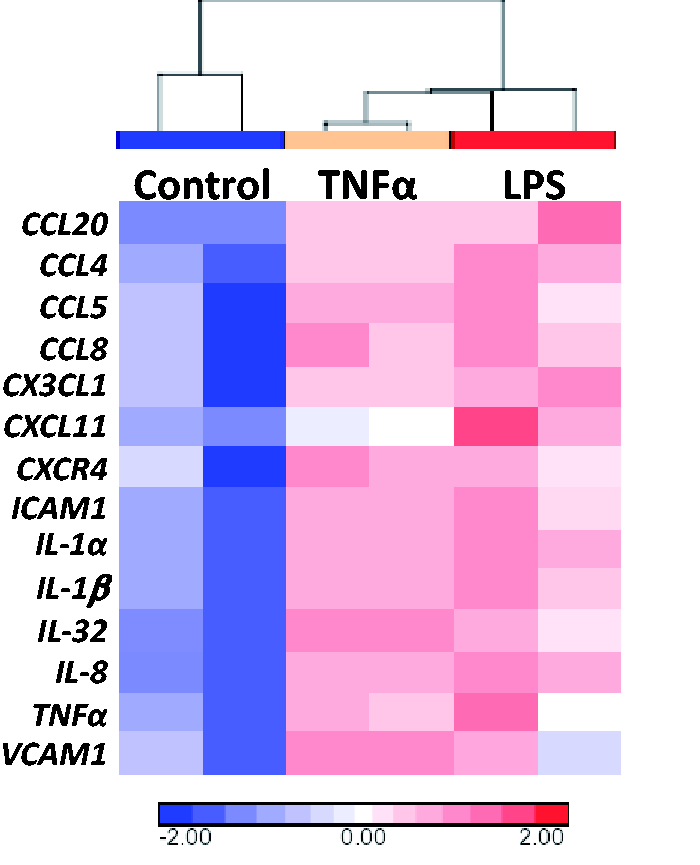
With respect to control genes for expression normalization, 18S rRNA is commonly used to assess basal transcription levels by qPCR and was initially chosen as an internal control to verify expression stability relative to GAPDH. Curiously, levels of 18S rRNA were observed to be highly variable in response to LPS in our initial experiments, with an overall down-regulation following stimulation with LPS (Figure 4B). Additional internal controls run to check for instability in housekeeping gene activity found this phenomenon to be exclusive to 18S rRNA, with levels of GUSB, TBP and GAPDH remaining stable after stimulation with either LPS or TNF-α (Figure 4C). rRNAs are structural subunits of ribosomes and have found extensive use as controls in gene expression studies,15,16 proving in certain instances to be even more robust (i.e. stable) than other commonly accepted housekeeping genes such as GAPDH or β-actin.17–19 In stark contrast to this, we observed that 18S rRNA levels were dramatically altered when cells were challenged with LPS. However, this is not an unprecedented finding. In a report published by Cai et al.,
20
it was observed that both 28S and 18S rRNA degradation occurs in response to LPS challenge in macrophages. Their study also examined levels of GAPDH and β-actin by nuclear run-on analysis and determined that transcription was unaltered, but that a global down-regulation of protein synthesis was observed as a result of rRNA degradation.
20
Furthermore, their work linked the effect of rRNA degradation to increased iNOS expression in macrophages following inflammatory cytokine production resulting in augmented NO production.
20
In order to follow this lead we also examined iNOS expression in our present work. However, our findings were unable to demonstrate augmented expression of iNOS in hPSC-derived airway epithelia, thus leading us to speculate that 18S rRNA degradation can occur in an NO-independent manner as well. Future studies will be needed to explore this observation in greater detail.
Changes in 18S rRNA levels following LPS challenge. (A) Melt curve for 18S rRNA PCR product and primer sequence. (B) Mean levels of 18S rRNA evaluated by qPCR following LPS challenge normalized to controls. 18S rRNA levels were significantly reduced in response to LPS. (C) Trace for fluorescence versus cycle number showing a large dynamic range of 18S rRNA expression levels, making it inappropriate for use as a control gene. The other control genes used in this study, GUSB, TBP and GAPDH, demonstrated tight amplification dynamics irrespective of LPS or TNF-α treatment, all plotted together. Fwd: forward; Rev: reverse; RFU: relative fluorescence units.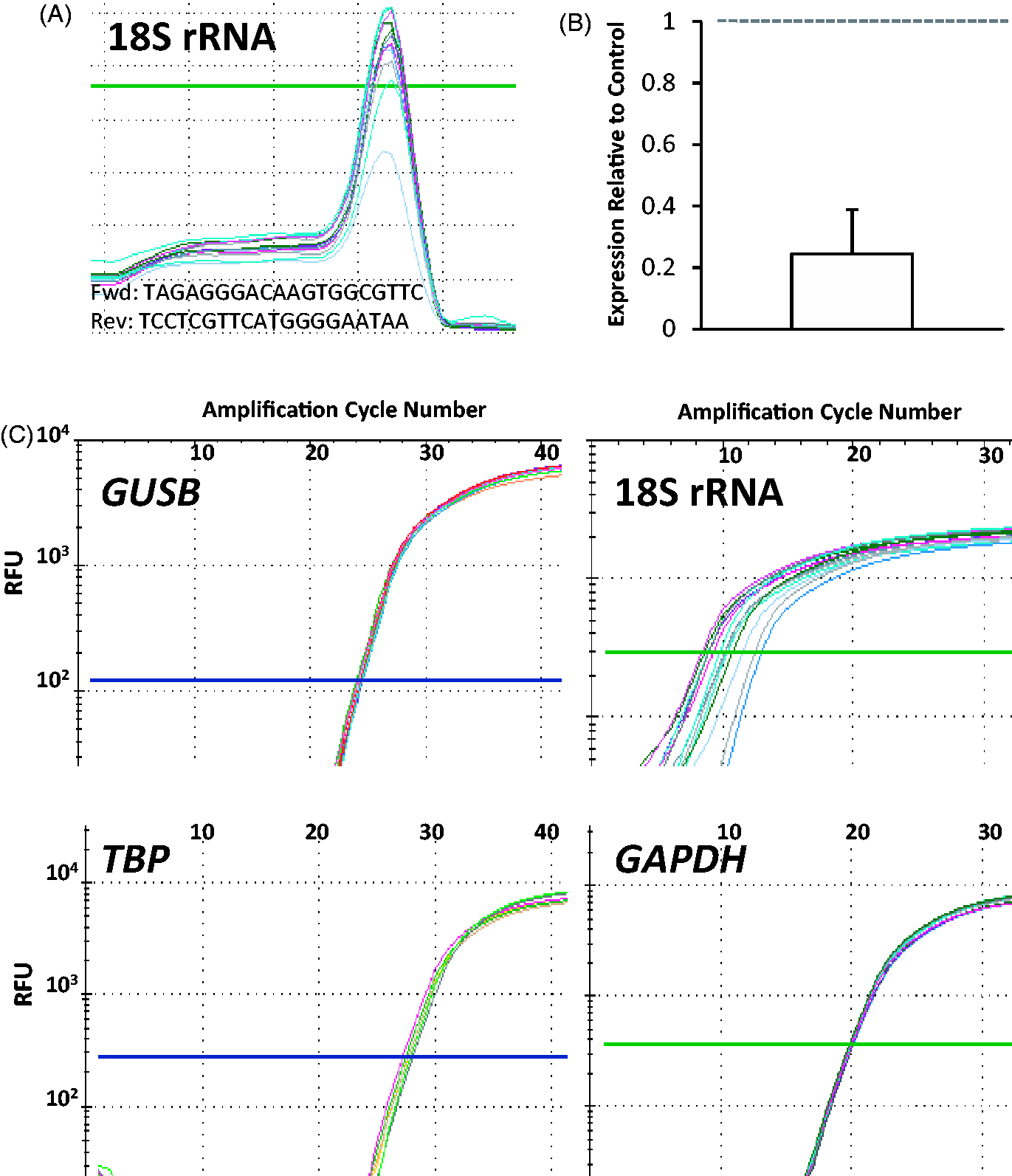
In addition to transcript measures, protein levels following TNF-α stimulation were also measured by ELISA (Figure 5). The results of the ELISA confirmed an increase in TNF-α protein production in response to LPS, correlating with the increase observed in the transcription of this gene, by 5 h (Figure 5A). Secreted TNF-α levels from cells stimulated with TNF-α had high levels of this protein beyond the accurate detection range of the assay (Figure 5A, B). This is to be expected, as the top value of the standard series is 500 pg/ml (Figure 5B), whereas we used 10 ng/ml of TNF-α in challenging airway epithelial cells generated from hPSCs, which can, theoretically, still be present in the collected conditioned apical supernatant.
TNF-α protein levels measured by ELISA. Samples treated with LPS had undetectable levels of TNF-α at 2 h. At 5 h post-LPS treatment TNF-α levels were >200 pg/ml. Stimulation with TNF-α, serving as a positive control for the assay. Inset: standard curve generated from a dilution series of TNF-α, demonstrating the upper limit of the assay at 500 pg/ml. Abs: absorbance.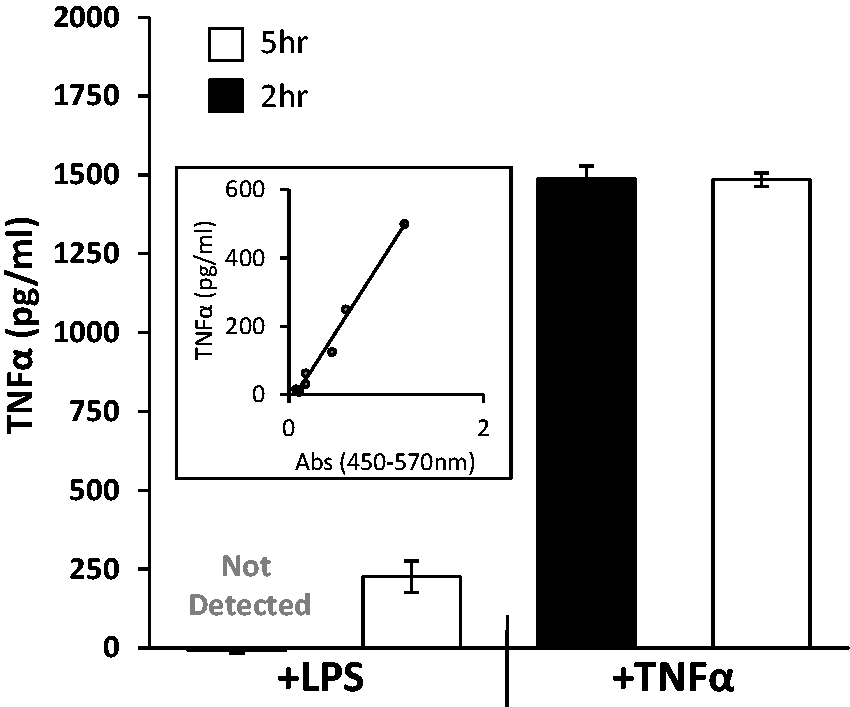
Discussion
Our study demonstrates a robust response of hPSC-derived airway cells to challenge with bacterial endotoxin and inflammatory cytokines. This response would appear to be mediated by airway epithelium and not other cell types, as genes that are specifically associated with non-airway cell types did not have any observable increase in transcription in response to challenge. The expression array data presented in this study, which has been uploaded to a public repository, will also allow the broader research community to cross-validate their findings with our model of airway-derived epithelium. To the best of our knowledge, this is the first time that the innate immune response of a hPSC-derived tissue has been evaluated.
In this study, lung epithelial genes were up-regulated following challenge with LPS and TNF-α, but other genes predicted to show increased expression levels, such as ESE1, did not. However, the study of Reddel et al., demonstrating that ESE1 is responsive to LPS and TNF-α challenge in airway epithelium, made use of a transformed human bronchiolar epithelial cell line, BEAS-2B. 21 There are two reasons why their findings do not translate to the present study. First, BEAS-2B is a bronchial epithelial cell line, and evidence from our work shows that hPSC-derived airway cells may be more representative of distal alveolar cells, 3 which have unknown ESE1 activity. Second, according to the ATCC (www.atcc.org), BEAS-2B has multiple karyotypic abnormalities, and confirmation of these findings were made in the lung carcinoma line A549, 22 which, according to the ATCC, has a modal chromosomal count of 66. In light of these observations it is tempting to theorize that primary cultures may not have the same order of response as observed in transformed, immortalized cultured lines.
Furthermore, the airway cells differentiated in our protocol may possess a cellular phenotype associated with a distinct spatial identity reflected by the proximal to distal hierarchy of the airway epithelium, which may thus explain why BD2 up-regulation was not observed. Studies demonstrating an increased transcription of BD2 in response to TNF-α examined bronchial, tracheal and nasal epithelia, but never alveolar epithelial cells. 11 These cell type-specific differences can also account for the lack of up-regulation of VEGFa, as its response was reported only in primary human nasal cells of unknown origin, termed generically as nasal fibroblasts by the authors, and not alveolar epithelial cells. 9
The generation of lung airway from hPSCs has potential broad implications in the field of regenerative medicine and basic experimental and developmental biology. Demonstrating the differentiation of airway cells with a functional innate immune response gives researchers a tool to dissect the response of, for instance, patient-derived hPSCs harboring genetic diseases predisposing their lungs to infection with pathogens as observed in patients with cystic fibrosis (CF). As infection with common bacteria such as Pseudomonas aeruginosa can pose a severe threat to patients with CF by colonizing the airways, CF-hPSCs that have been differentiated into airway cells can be used in a screening setting to identify novel drugs useful in eliminating pathogens or, more importantly, to boost the innate immune response for the clearance of pathogens; yet, robust bacterial/human airway epithelial co-culture systems would first need to be adapted based on their more widespread use in modeling gastrointestinal tract host–pathogen interactions.23,24
Innate immune activation in airway epithelial cells serves as the first step for immune recognition, which transcends into production of cytokines/chemokines that drive dendritic cell migration to the draining lymph nodes, resulting in priming and initiation of a T-cell response. 25 As such, innate immune activation of epithelial cells has been implicated in mediating pulmonary pathology in the context of airway infections, autoimmune disorders, such as asthma, and genetic diseases such as CF. Inflammasome activation in epithelial cells by viral infection leads to production of cytokines such as IL-1β and IL-8, 26 which play a role in driving pulmonary dendritic cell migration to the draining lymph nodes to initiate an antiviral immune response. Moreover, recognition of viral particles by airway epithelial cells also induces an IFN response, which plays a key role in limiting viral burden. 26 Furthermore, epithelial cells also produce cytokines such as thymic stromal lymphopoeitin, which drive a T-helper cell 2 (Th2) response, playing a major role in initiation and progression of an autoimmune response in airway diseases such as allergy and asthma.27,28 In patients with CF, airway epithelial cells express receptor for advanced glycation end products (RAGE), a versatile sensor of damage-associated molecular patterns, which is activated under conditions of hypoxia, as well as infections, and plays a role in mediating inflammation in the CF lung. 29 Therefore, modulation of immune activation in airway epithelial cells may hold the key for regulation of airway inflammation in different pathologies. As such, hPSC-derived airway epithelium offers a unique model to screen for molecules which can increase IFN response and inflammasome activation desired in antiviral response(s), suppress the production of Th2-driving cytokines desired in allergy and asthma, and even suppress RAGE activation, desired in suppressing CF lung inflammation.
Taken together, our findings demonstrate the functionality of airway epithelium generated from hPSCs in vitro. The ability to mount an innate immune response, a hallmark of functional airway epithelium, demonstrates the utility of this model for drug screening studies aimed at clinical application, in addition to providing an ethical surrogate to interrogate the basic biology of the development of innate immunity in human lungs. In conclusion, we propose that innate immune challenge provides a reliable, robust experimental benchmark to examine the differentiation of functional airway epithelium specifically from hPSCs. This field, which has relied on simple phenotyping experiments such as immunofluorescence and qPCR is in need of rapid functional assays, such as innate immune challenge, to assess differentiation.30,31 This assay can be either carried out by the collection of apical supernatant to measure protein concentrations by ELISA, or by sacrificing one well of an experiment for RNA extraction and qPCR analysis following LPS challenge. Providing this type of functional validation will give confidence moving forward in future downstream translational experiments involving preclinical animal models.
Footnotes
Funding
This work was supported by a CIHR Emerging Team Grant to M.B.; R.K. was supported by CIHR-Banting Fellowship.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.