
Abstract
Kinins are pro-inflammatory peptides that mimic the cardinal features of inflammation. We examined the concept that expression levels of endothelial intercellular adhesion molecule-1 (ICAM-1) and neutrophil integrins Mac-1 and LFA-1 are modulated by the kinin B1 receptor (B1R) agonist, Lys-des[Arg9]bradykinin (LDBK). Stimulation of endothelial cells with LDBK increased the levels of ICAM-1 mRNA transcripts/protein, and also of E-selectin and platelet endothelial adhesion molecule-1. ICAM-1 levels increased in a magnitude comparable with that produced by TNF-α. This stimulatory effect was reduced when endothelial cells, which had been previously transfected with a B1R small interfering RNA, were stimulated with LDBK, under comparable conditions. Similarly, LDBK produced a significant increase in protein levels of LFA-1 and Mac-1 integrins in human neutrophils, an effect that was reversed by pretreatment of cells with 10 µg/ml cycloheximide or a B1R antagonist. Functional experiments performed with post-confluent monolayers of endothelial cells stimulated with LDBK and neutrophils primed with TNF-α, and vice versa, resulted in enhanced adhesiveness between both cells. Neutralizing Abs to ICAM-1 and Mac-1 reduced the adhesion between them. Our results indicate that kinin B1R is a novel modulator that promotes adhesion of leukocytes to endothelial cells, critically enhancing the movement of neutrophils from the circulation to sites of inflammation.
Introduction
Migration of neutrophils from the vascular compartment into injured tissue sites is a hallmark of acute inflammation. In such events, cellular and molecular interactions occur between specific membrane proteins that reside on the surface of both endothelial cells and leukocytes. Endothelial cells express a family of major intercellular adhesion molecules known as intercellular adhesion molecule (ICAM)-1 and ICAM-2 (junctional adhesion molecules) and platelet endothelial adhesion molecule-1 (PECAM-1), which are implicated in endothelium–leukocyte interactions. Adhesion molecules are members of the Ig superfamily that contain one or more extracellular copies of the Ig folds, each of which is formed by a 70–110-amino acid sequence held together by two cysteine residues at their N- and C-termini. 1 These adhesion molecules comprise a large N-terminal extracellular domain containing the Ig folds, a single transmembrane helical domain and a cytoplasmic tail. 2 ICAM-1 is constitutively expressed uniformly, but at low levels on the surface of endothelial cells. Expression of ICAM-1 is up-regulated after contact with inflammatory cytokines such as IL-1 and TNF-α.3,4
The most important leukocyte ligands that interact with ICAM-1 exposed on the endothelial cell surface are the integrins, Mac-1 (CD11b/CD18) and LFA-1 (CD11a/CD18), which bind to separate Ig domains of ICAM-1 together with the simultaneous binding of both integrins to one ICAM-1 molecule. 2 Interaction between these integrins and ICAM-1 has been demonstrated both in vitro and in vivo and results, to a varying degree, in the transmigration of leukocytes through endothelial cells when stimulated by cytokines or leukocyte chemoattractants. 4 Importance of the integrin–ICAM-1 interaction is supported by the fact that ICAM-1 knockout mice have impaired inflammatory and immune responses characterized by reduced leukocyte migration into inflamed tissues. 5
Cellular mediators trigger changes that occur in the microcirculation of inflamed tissue. Prominent among them are the peptide hormones known as kinins, generated by the enzymic action of either plasma kallikrein or tissue kallikrein (KLK1) on endogenous substrates called L- and H-kininogens. Evidence supports a major role for the kinin peptides in an acute inflammatory response.6,7 Injections of bradykinin mimic the cardinal features of inflammation, namely erythema, edema (increase in vascular permeability) and pain. The local acid microenvironment that prevails in inflamed tissue is favorable for the accumulation of kinins, 6 and components of the kallikrein–kinin cascade have been identified at such sites. Neutrophils that migrate to inflamed sites play a pivotal role in the formation of kinins as they possess all the components of the kallikrein–kinin cascade, namely membrane bound kininogens,8,9 kinin-releasing enzyme, KLK110,11 and kinin B1 (B1R) receptor. 12 In fact, stimulation of human neutrophils with the chemotactic peptide fMet–Leu–Phe (fMLP) or opsonized silica particles releases KLK1 that, upon contact with kininogens, produces biologically active kinin peptides. 13 Clearly, the neutrophil is fully equipped to form kinin peptides de novo, suggesting that its kinin cascade may function in an autocrine/paracrine manner. Similar to neutrophils, endothelial cells express B1 and B2 receptors and express or bind kininogens, the endogenous substrates of the kinin system. 14 Activation of kinin receptors on endothelial cells may result in arteriolar vasodilatation, release of nitric oxide and PGI2 and increase in vascular permeability.15–17 Injection of a B1R agonist into a mouse causes a dose-related increase in vascular permeability, which is accompanied by neutrophil influx, a response that seems to be mediated by the release of neuropeptides and nitric oxide. 18 In support of the concept that B1R participates in the recruitment of neutrophils is the observation that when peritonitis is produced experimentally in B1R knockout mice the number of neutrophils that migrate into the inflammatory milieu is significantly reduced. 19 Furthermore, our recent studies show that activation of B1R in human neutrophils initiates chemotaxis and triggers signaling cascades that result in the release of components from tertiary and primary granules.12,20 In addition, B1 and B2 receptor agonists have been shown to increase leukocyte adhesiveness to components of the extracellular matrix such as fibrinogen and fibronectin, 21 and also induce leukocyte migration both in vitro and in vivo.22,23
We therefore designed experiments to investigate the interaction between neutrophils and endothelial cells by determining whether this dynamic event was modulated by kinin peptides. Our first aim was to determine the expression levels of integrins, LFA-1 and Mac-1 on human neutrophils and those of the intercellular adhesion molecules in endothelial cells after stimulation of kinin B1 receptors. The second aim was to examine the question whether Mac-1 and ICAM-1 achieved contact between one another in an in vitro assay that used a post-confluent monolayer of endothelial cells and neutrophils subsequent to stimulation by a natural B1R agonist.
Materials and methods
Ethical permission
We followed the guidelines stipulated by the Medical Ethical Committee of Universidad Austral de Chile and the Declaration of Helsinki principles. Blood was collected by venepuncture from 10 healthy volunteers (six men, four women), ranging in age from 23 to 38 yr. All had a body mass index < 27 kg/m2. Any volunteer taking chronic medication, suffering from a cold, or having an allergic or any other inflammatory disorder was excluded from the study.
Isolation of human neutrophils
Blood (35 ml) collected in 50-ml sterile tubes containing 3.8% w/v trisodium citrate, was mixed with 35 ml of dextran (6% w/v, average molecular mass 500 ku; Sigma Aldrich, St. Louis, MO, USA) and 105 ml of PBS (10 mM sodium phosphate, 2.7 mM KCl and 137 mM NaCl, pH 7.4) containing 0.4% w/v trisodium citrate. After 20 min at room temperature (20–22℃), the upper leukocyte-enriched plasma was centrifuged and the cell pellet resuspended in 1.5 ml of a 50% Percoll (Sigma Aldrich) solution that was placed onto a Percoll gradient to separate neutrophils, as described previously. 9 Cell viability, assessed by Trypan Blue exclusion, was 99%. All procedures were carried out under sterile conditions.
Flow cytometry
Aliquots (100 µl) of heparinized whole blood were incubated directly with different concentrations (1–100 nM) of fMLP or the B1R agonist Lys-des[Arg9]bradykinin (LDBK; Sigma) for variable periods of time (15–180 min). After lysis of red cells, leukocytes were stained for surface markers with APC mouse anti-human CD11a or CD11b (BD Biosciences, San Jose, CA, USA). Next, immunolabeled leukocytes were analyzed using a FACSCanto II flow cytometer with FACSDiva version 6.1.1 and FlowJo 7.6 software (BD Biosciences). Initially, a gate was placed in forward scatter/side to exclude cell debris. The population of neutrophils was identified using a forward scatter versus side scatter plot to determine the relative size and granularity of the cells. Thereafter, the mean intensity fluorescence of neutrophils expressing each surface-labeled integrin was gated and analyzed. Values obtained after stimulation with fMLP or LDBK were compared with those obtained from non-stimulated controls.
Cell culture
The human endothelial cell line EA.hy926 was a gift from Dr. Cora-Jean S Edgell (Department of Pathology, University of North Carolina at Chapel Hill, NC, USA). Cells were cultured at 37℃ using DMEM supplemented with 100 mM hypoxanthine, 0.4 mM aminopterin and 16 mM thymidine (Invitrogen, Grand Island NY, USA), 10% FBS (Invitrogen), 200 mM glutamine and a mix of antibiotics and fungicide (Sigma Aldrich), hereafter called DMEM-HAT.
RT-PCR
List of primers, sequences and their product size to amplify B1R, ICAM-1 and house-keeping genes by semiquantitative and quantitative PCR.

Changes in ICAM-1 expression were evaluated by quantitative PCR on cDNA of endothelial cells using a Mx3000P® thermocycler (Stratagene, Wilmington, DE, USA), Brilliant II SYBR Green qPCR Master Mix (Stratagene), according to manufacturer's instructions, and the primer pairs for ICAM-1 25 and β-actin 26 (Table 1). The settings for the melting curve protocol were as follows: denaturation at 95℃, cooling to 60℃ (5℃ above the primer annealing temperature) and heating to 95℃ (speed 0.1℃/s). The fluorescence emitted by double-stranded DNA-bound SYBR-Green was measured once at the end of each additional heating step, and continuously during the melting curve program. The presence of one product was confirmed by melting curve analysis and product size. Efficiency for ICAM-1 and β-actin was 103.1% and 98.3%, respectively. The results obtained were normalized with respect to β-actin expression considering the efficiency recorded for each gene and expressed as fold changes with respect to the unstimulated group.
Western blotting
Stimulation of isolated neutrophils or synchronized endothelial cells with B1R agonist was stopped by the addition of 100 µl of ice-cold RIPA buffer, and cells were sonicated and the amount of protein measured. 20 Equal amounts of protein, dissolved in sample buffer containing 2.5% mercaptoethanol, were subjected to SDS-PAGE and then electrotransferred onto immobilon P. Blots were incubated for 2 h with primary Abs to LFA-1, Mac-1 and ICAM-1 (AbCam, Cambridge, UK) in PBS containing 0.1% Tween 20 and 5% BSA. Bound Abs were detected using a chemiluminescence kit (Pierce, Rockford, IL, USA). The total amount of protein present in each sample was assessed on the same membrane using polyclonal Abs to actin or GAPDH. For this purpose, the Abs used for the first immunodetection were stripped previously for 30 min at 50℃ with 62.5 mM Tris-HCl pH7.4 containing 100 mM β-mercaptoethanol and 2% SDS.
Effect of kinin receptor antagonists, and inhibitors
To establish the specificity of the activated kinin receptor, neutrophils or endothelial cells were pre-incubated for 30 min with an excess of either B1R antagonists des[Arg9]-Leu8-BK (Sigma) or Lys-des[Arg9]-Leu8-bradykinin (Bachem, Torrance, CA, USA), or the B2 receptor antagonist HOE140 (Aventis Pharma Deutschland GmbH, Frankfurt am Main, Germany). The B1R agonist and the antagonists were suspended in physiologic sterile 0.9% NaCl, whereas DMSO was used to dissolve all inhibitors. Both, ligands and inhibitors were additionally diluted with HBSS or DMEM at the beginning of each experiment. To determine expression of the integrins Mac-1 and LFA-1 on neutrophils, they were treated with cycloheximide (1 or 10 µg/ml) or brefeldin A (10 µg/ml) for 60 min before stimulation with agonist. Controls included the replacement of the inhibitor by the corresponding vehicle.
Transfection of B1R small interfering RNA
Endothelial cells were transfected as described previously. 27 To summarize, confluent monolayers (1 × 106 cells) were placed in DMEM without antibiotics and antimicotics, and incubated with small interfering RNA (siRNA)–lipofectamine complexes prepared according to the Lipofectamine'2000 protocol (Invitrogen). Transfection was performed for 24 h in the same medium with 100 pmol of B1R siRNA (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or control siRNA (Block-it fluorescent oligo; Invitrogen). Efficiency of transfection was controlled 24 h and 48 h after transfection by detecting the incorporated block-it fluorescent oligo by fluorescence microscopy. Silencing of the B1R, after using this protocol was determined at the level of the expression of B1R protein and mRNA, and phosphorylation of ERK1/2 MAPK in transfected cells stimulated with LDBK (not shown). Prior to stimulation with the B1R agonist cells were incubated for an additional period of 24 h in DMEM with antibiotics and antimicotics.
Immunocytochemistry
Endothelial cells were grown in DMEM supplemented with 10% FBS on 35-mm tissue culture dishes containing a 22 × 22 mm coverslip. After washing with Dulbecco's PBS (DPBS) cells were fixed with methanol at −20℃ for 45 min, completely air dried and stored at −80℃ until used. For immunolabeling, coverslips were incubated with DPBS containing 3% BSA at room temperature for 30 min to prevent non-specific labeling and then incubated overnight at 4℃ with anti-B1R Abs or a monoclonal Ab against ICAM-1 (1:100) in DPBS-1% BSA. Two Abs directed to specific peptide sequences of the B1R were used. One Ab (1:500) was raised against a peptide (ISSSHRKEIFQLFWRN) that corresponded to the C-terminal end of kinin B1R, kindly provided by Merck & Co (West Point, PA, USA),12,28 whereas the other corresponded to a mixture (1:250 to 1:500) of three antisera directed to peptide sequences of extracellular domains 1 (ELQSSNQSQLFPQNATACDNAPEAWDLLHR), 2 (ENIWNQFNWPFGALLCR) and 3 (RSIQAVPDLNITACILLLPHEAWHFARIVE) of kinin B1R, kindly donated by Prof. W Müller-Esterl (Goethe University, Frankfurt, Germany). 29 In addition, live endothelial cells were incubated for 4 h on ice with the mix of antisera directed to extracellular domains 1–3 and then fixed with absolute methanol. Next, cells were incubated with an anti-mouse F(ab')2 fragments conjugated to Alexa 488 (1:1000; Molecular Probes, Eugene, OR, USA). Nuclei were detected with 0.5 µg/ml DAPI in DPBS. Immunolabeling was evaluated in an Olympus Fluoview FV1000 confocal microscope using the 488-nm lane of Argon-Krypton and 405 nm of diode lasers to the excitation of Alexa 488 and DAPI, respectively. Optical sections taken every 0.4 µm were digitalized and images processed using Image J software (NIH, Bethesda, MD, USA). Negative controls included omission of primary Ab or its replacement by non-immune serum.
Neutrophil–endothelial cell adhesion assay
Post-confluent cultures of endothelial cells were grown in 24-well plates and synchronized overnight in the absence of FBS. In the first set of experiments, endothelial cells were stimulated with 1 nM TNF-α for 6 h and neutrophils with varying concentrations of LDBK (1–100 nM) for 30 min. After this period, neutrophils were washed and co-incubated with endothelial cells for 30 min at 37℃, and then rigorously washed. Adhesion of neutrophils to the monolayer was evaluated my measuring myeloperoxidase activity in extracts prepared with RIPA buffer without inhibitors. In the second set of experiments the agonists were reversed, endothelial cells were stimulated with LDBK and neutrophils with 1 nM TNF-α, and the same technical procedure repeated as described above. Positive controls were performed with both endothelial cells and neutrophils stimulated with TNF-α. The following negative controls were designed: (i) both cell types remained unstimulated, (ii) either endothelial cells or neutrophils were stimulated with TNF-α, (iii) cells were pretreated with a B1R antagonist prior to stimulation with LDBK, (iv) neutralizing Abs to Mac-1 (5–10 µg/ml; BD Biosciences) or to ICAM-1 (1:100; Dako, Carpinteria, CA, USA) were added in the assay. Each experiment was performed in triplicate and the mean ± SEM of five independent experiments was plotted.
Quantitative image analysis and statistics
Intensity of immunoreactive protein bands, after immunoblotting, was quantified by an automated image digitizing system (Un-Scan-It; Silk Scientific Inc., Orem, UT, USA), as described previously. 12 At least three independent experiments were performed for each measurement and representative images from different experiments are depicted in the figures. Values are expressed as mean ± SEM and significance was considered acceptable at the 5% level (P < 0.05). Paired or unpaired Student's t-test was used for comparison between groups.
Results
Kinin B1R stimulation increases integrin levels on human neutrophils
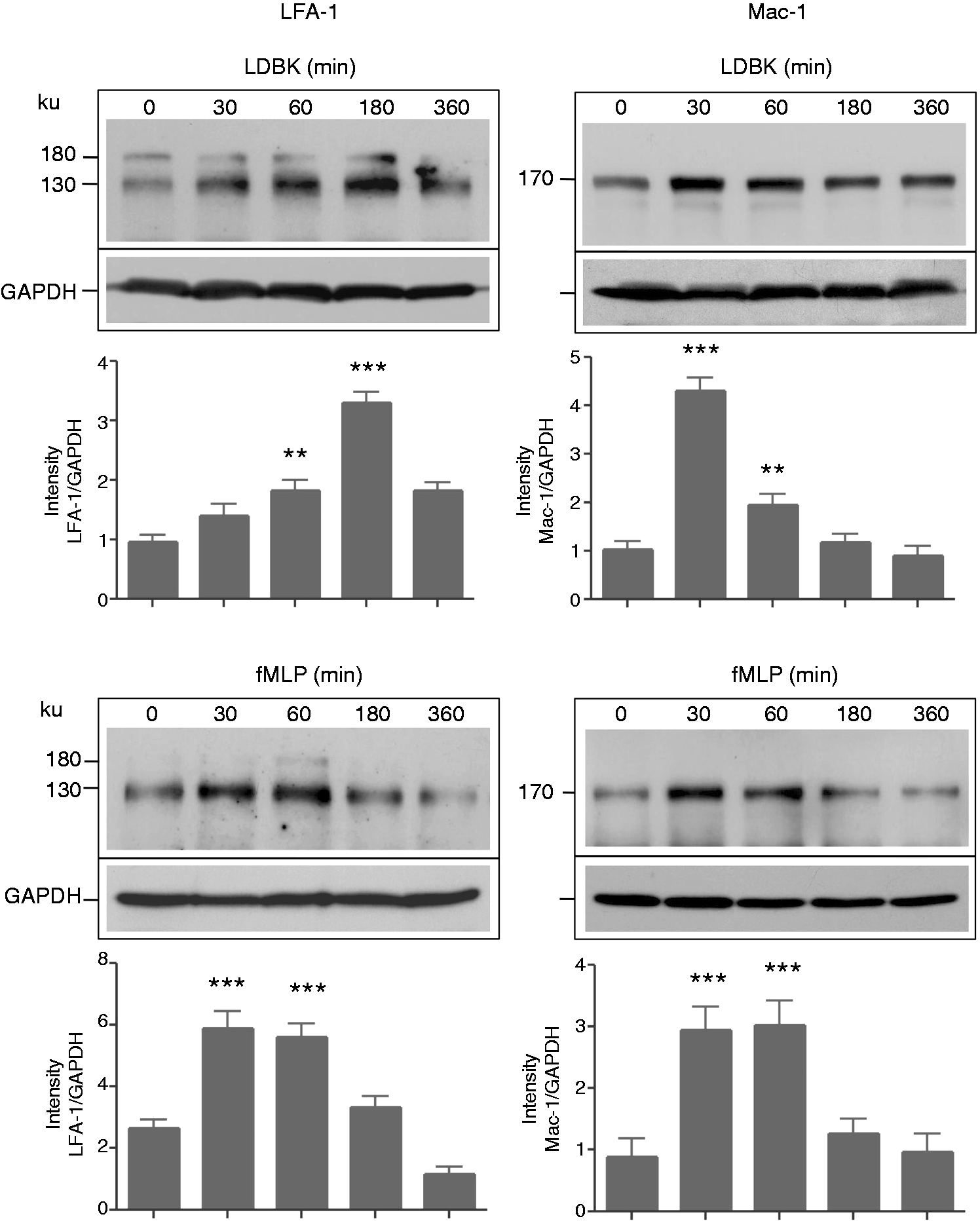
Our first approach was to determine the effect of activating neutrophil kinin B1R on the two integrins, LFA-1 and Mac-1, by adding either the B1R agonist, LDBK or the chemotactic peptide fMLP (positive control) to aliquots of whole human blood and evaluating the response by flow cytometry. When aliquots were incubated with 100 nM LDBK or 1 µM fMLP for 15 min, the amount of Mac-1 on the neutrophil cell membrane was increased when compared with non-stimulated cells; the increase was more pronounced after 3 h of incubation with LDBK (Figure 1). Stimulation of whole blood with 100 nM LDBK translocated Mac-1 to the neutrophil cell membrane in an amount that was approximately 20% of the integrin translocated by 1 µM fMLP. In contrast, levels of LFA-1 were unchanged after 15 min of stimulation by LDBK or fMLP, and changed only partially when the samples were incubated for 3 h (Figure 1). Time course experiments in which both integrins were evaluated in homogenates of purified neutrophils stimulated with 100 nM LDBK showed that total LFA-1 levels (membrane-bound and cytoplasmic integrins) had increased between the 60 and 180 min period of stimulation with a maximal increase occurring after 180 min of stimulation (Figure 2). LFA-1 displayed an expected molecular mass of 130 ku, but, in addition, there was an extra immunoreactive band of 180 ku formed as a result of post-translational modification, for example glycosylation that increases the molecular mass of proteins. For data analysis, the 130-ku band was considered to represent the amount of immunoreactive protein in each sample. Total Mac-1 reached its highest levels after 30 min of stimulation with LDBK (Figure 2). By comparison, neutrophils stimulated with fMLP under identical conditions showed augmented levels for both integrins during a period of 30–60 min post-stimulation (Figure 2). Importantly, in dose-dependent experiments, levels of Mac-1 increased significantly in neutrophils stimulated for 30 min at concentrations of B1R agonist that ranged between 10 to 100 nM, whereas levels of LFA-1 increased significantly only when neutrophils were stimulated for 180 min with 1–100 nM LDBK (Figure 3A).
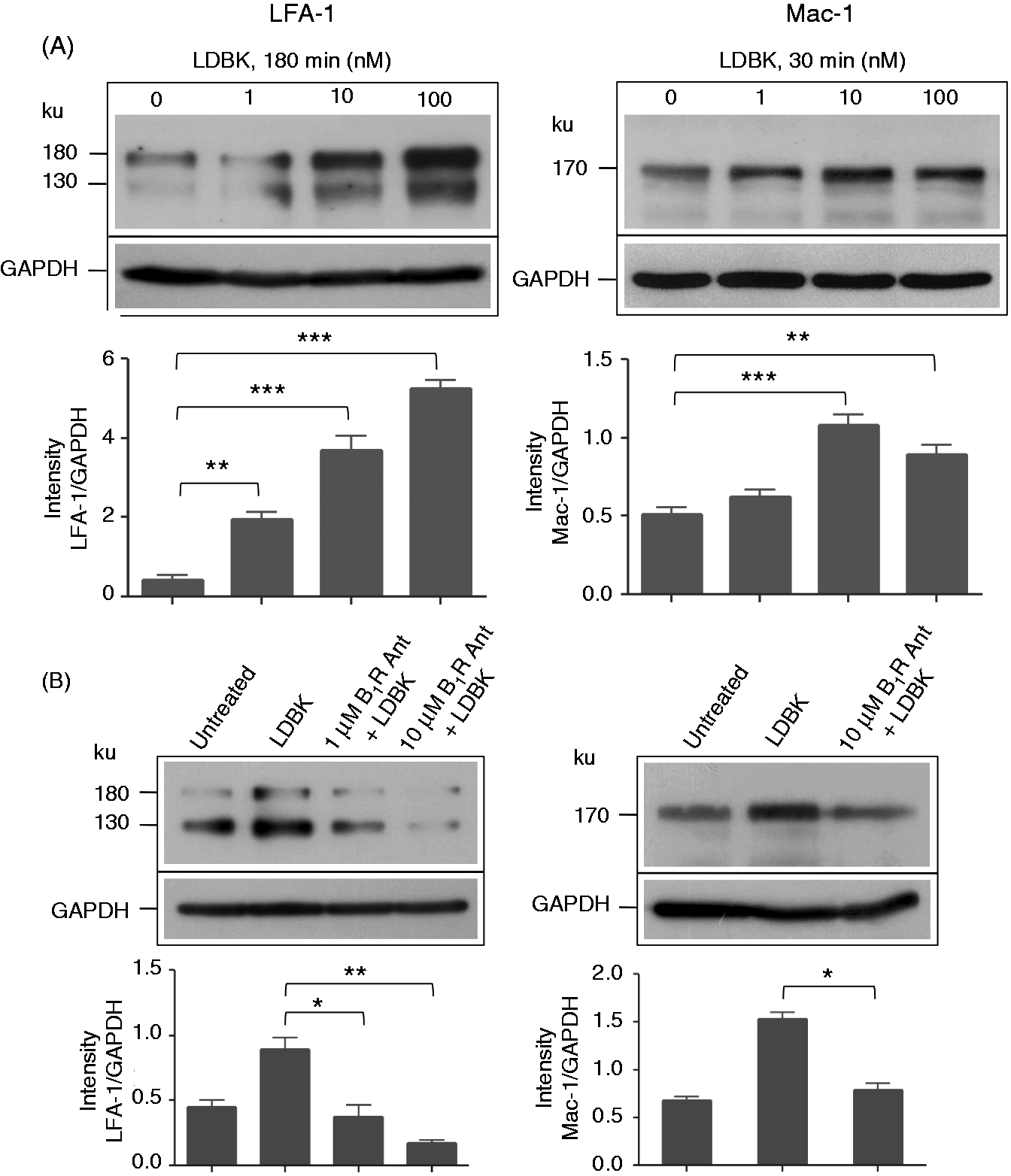
Identification of Mac-1 and LFA-1 integrins in human neutrophils by flow cytometry. Aliquots of whole human blood were incubated with the B1R agonist, LDBK for 15 min or 3 h. Then red cells were lysed, and white cells fixed and incubated with anti-Mac-1 Abs for 1 h. Bound Igs were detected using APC-labeled anti-mouse IgG for 30 min. Positive controls were performed using 1 µM fMet-Leu-Phe (fMLP). The figure was prepared with the FlowJo 7.6 software and is a representative presentation of three independent experiments. Levels of Mac-1 and LFA-1 integrins in neutrophils stimulated with LDBK or fMLP for various periods of time. Neutrophils were stimulated with 100 nM LDBK or 100 nM fMLP (fMet–Leu–Phe), and proteins separated by SDS-PAGE and transferred onto Immobilon P and then immunoprinted for Mac-1 and LFA-1. Representative Western blots of three independent experiments (n = 3) are shown. **P < 0.01; ***P < 0.001. Levels of Mac-1 and LFA-1 integrins in neutrophils stimulated with LDBK in the absence or presence of a B1R antagonist. (A) Neutrophils were stimulated with various concentrations of LDBK for 30 min (Mac-1) or 180 min (LFA-1), proteins were separated by SDS-PAGE, and Mac-1 and LFA-1 visualized by Western blotting. (B) Cells were stimulated with 10 nM LDBK after pre-incubation for 30 min with the B1R antagonist (B1R Ant) desArg9[Leu8]-bradykinin. Data represent the mean ± SEM of three independent experiments (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001.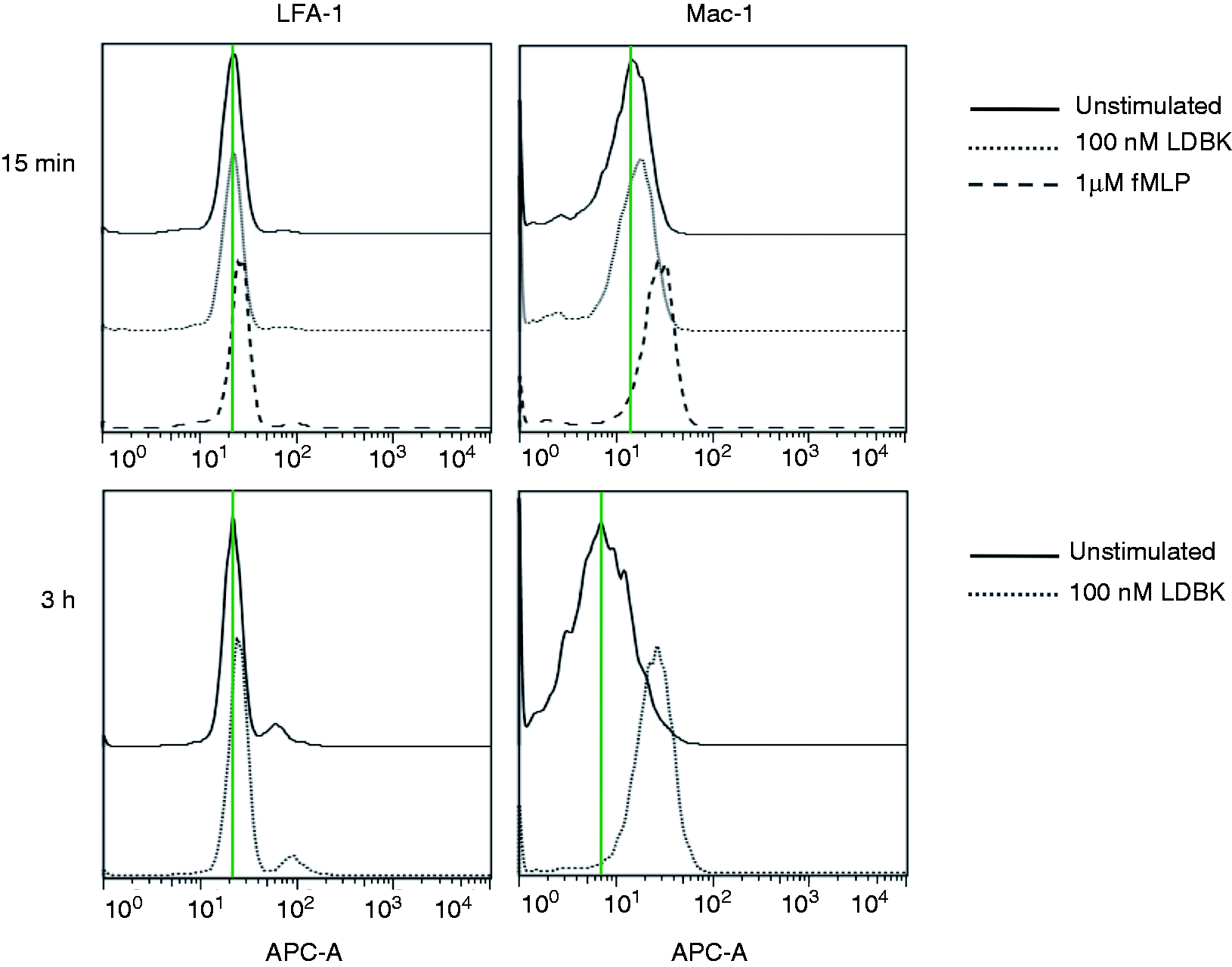
Increase in the integrins levels was inhibited by B1R antagonists and reduced by cycloheximide
Pretreatment of human neutrophils with the kinin B1R antagonists, desArg9[Leu8]-bradykinin or Lys-desArg9[Leu8]-bradykinin (1 or 10 µM) prior to stimulation with 10 nM LDBK entirely blocked an increase in the expression of LFA-1 and Mac-1 (Figure 3B). The effect of B1R antagonists indicated that LDBK had enhanced expression of neutrophil integrins by activating kinin B1R.
The next question we addressed was whether the increase in LFA-1 and Mac-1 depended on de novo synthesis triggered by stimulation of B1R. LDBK stimulation of isolated neutrophils pretreated with cycloheximide resulted in a dramatic decrease in the expression of LFA-1 (Figure 4A). Similarly, expression of Mac-1 was significantly reduced after pretreatment of leukocytes with 10 µg/ml cycloheximide (Figure 4A). Additional evidence for the hypothesis that activation of B1R modulated synthesis of integrins was provided by parallel experiments in which the pretreatment of neutrophils with brefeldin A significantly increased the content of LFA-1 and Mac-1 (Figure 4B). In control experiments, no change in the expression of integrins was observed in neutrophils incubated with solvents used to suspend the agonist or antagonists (0.9% NaCl) or inhibitors (DMSO).
Effect of cycloheximide and brefeldin A on Mac-1 and LFA-1 levels. Neutrophils were stimulated with 10 nM LDBK after pre-incubation for 60 min with (A) cycloheximide (CHX) or (B) brefeldin A (BFA). Cycloheximide or brefeldin A by themselves do not affect the levels of Mac-1 or LFA-1. Data represent the mean ± SEM of three independent experiments (n = 3). *P < 0.05; **P < 0.01 between stimulated cells and those pretreated with cycloheximide or brefeldin A prior stimulation with LDBK. #P < 0.05 between untreated neutrophils and those stimulated with LDBK.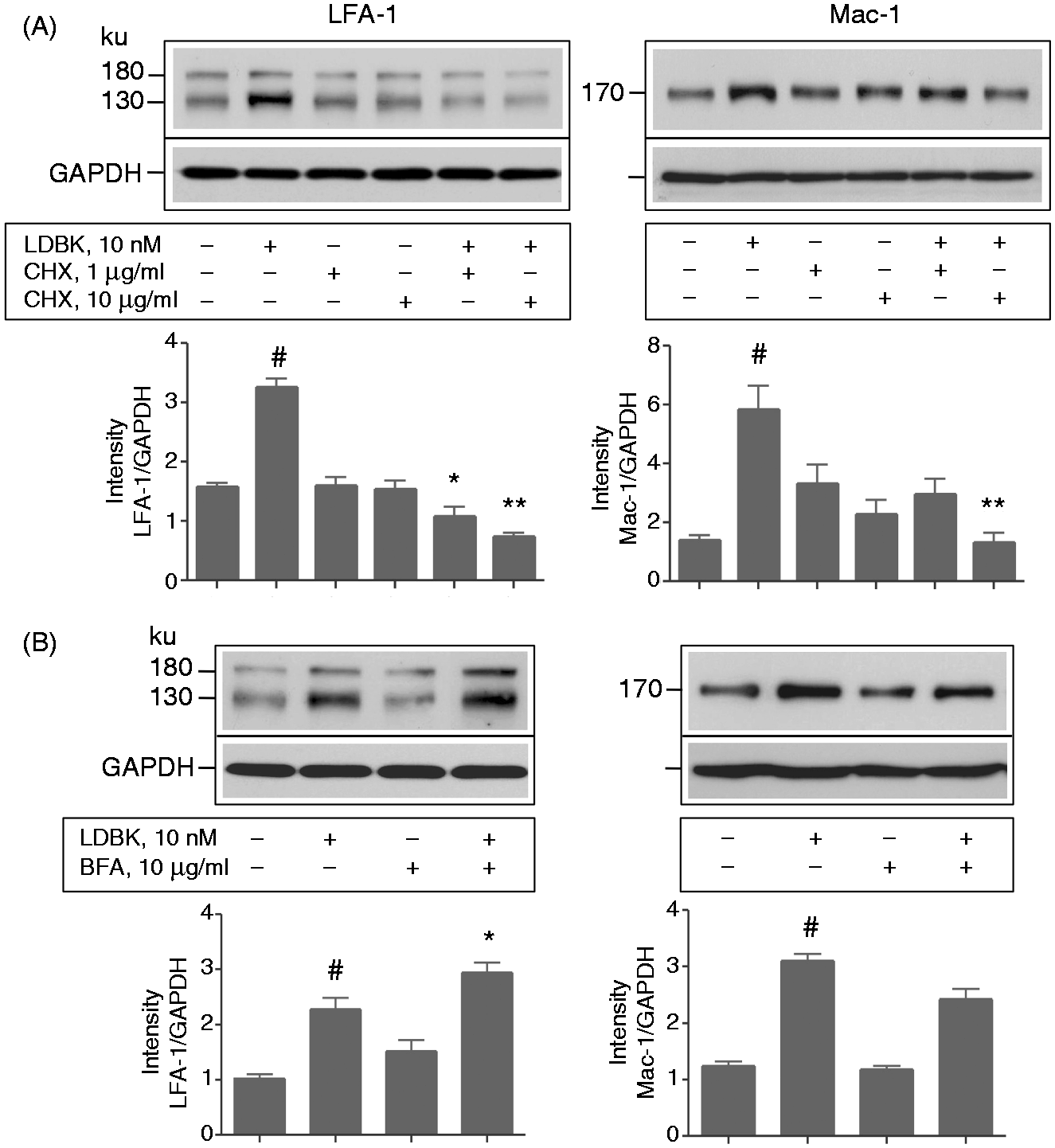
Kinin B1R stimulation induces expression of intercellular adhesion molecules on human endothelial cells
Functionally, the intercellular adhesion molecules of endothelial cells appear to mirror the integrins of neutrophils. This analogy prompted us to investigate whether stimulation of human endothelial cells by a B1R agonist would cause changes in the expression of intercellular adhesion molecules. However, in the absence of proof for the presence of B1R in the human endothelial cell line EA.hy296, basal experiments were designed to identify both the B1R mRNA and protein. Experiments using RT-PCR, Western blotting and immunocytochemistry clearly demonstrated the presence of B1R in this endothelial cell line (Figure 5). Furthermore, endothelial cells expressed the tyrosine kinase VEGFR-2 receptor, which characterizes human endothelial cells (Figure 5A). When membrane fractions from subconfluent endothelial cell cultures were subjected to Western blotting with an antiserum to the human B1R, two major bands of 45 and 35 ku were observed, similar to those described for B1R in human neutrophils
12
and breast cancer cells (Figure 5B).
30
The classical description of the up-regulation of B1R by cytokines such as TNF-α was confirmed by using membrane fractions prepared from stimulated and non-stimulated endothelial cells (Figure 5B). Silencing of B1R with a specific siRNA reduced the amount of B1R protein in membrane fractions when analyzed by Western blots (Figure 5D). Non-immune serum applied under the same conditions failed to produce significant immunolabeling of the membrane fractions (not shown). When the antisera were utilized for immunolabeling experiments B1R was visualized in the cytoplasm as well as on the plasma membrane of endothelial cells, especially when the mix of antiserum directed to extracellular domains 1–3 was used on live cells at 4℃ (Figure 5C). Omission of B1R antiserum or co-incubation of cells with an excess of the same peptide used for immunization resulted in absence of immunostaining (Figure 5C). To further emphasize the expression of a functional B1R on endothelial cells we determined the phosphorylation of ERK1/2 MAPK, a signaling pathway associated with up-regulation of intercellular adhesion molecules, in response to B1R stimulation. As anticipated, a rapid phosphorylation of ERK1/2 that lasted up to 30 min occurred when stimulated by LDBK in a range spanning 1 to 100 nM (Figures 5E, F). Phosphorylation was reduced markedly when the cells were pretreated with a B1R antagonist, but not with a B2R antagonist (HOE140), indicating that phosphorylation of ERK1/2 was produced by activation of B1R (Figure 5G).
Human EA.hy926 endothelial cells express the kinin B1R. Cells were cultured to subconfluency and then used to show expression and activity of B1R. (A) RT-PCR. (B) Stimulation with 1 nM TNF-α for 12 h shows the characteristic up-regulation of B1R in response to this cytokine. Membrane proteins were separated by SDS-PAGE, transferred onto Immobilon P and then immunoprinted with an antiserum raised against the C-terminus end of B1R. (C) Immunovisualization of B1R using the same antiserum or a mix of antisera directed to extracellular domains 1, 2 and 3 (ED1-3) of B1R. Negative controls were performed using pre-immune serum. (D) Cells were transfected with a B1R siRNA for 48 h and then B1R protein analyzed as described in (B). **P < 0.01 between siRNA-treated and non-treated cells. (E, F) Cells were incubated with various concentrations of LDBK for 15 min (E) or with 10 nM LDBK for different periods of time (F). Proteins were separated by SDS-PAGE as described above and immunoblotted with anti-phosphorylated ERK1/2 MAPK Abs (P-ERK1/2). Abs were stripped and Immobilon incubated with an Ab that recognizes phosphorylated and non-phosphorylated ERK1/2 (T-ERK1/2). *P < 0.05; **P < 0.01 between stimulated and non-stimulated cells. (G) Cells were pre-incubated with 1 µM of the B1R antagonist des[Arg9]Leu8-bradykinin (B1R Ant) or the B2R antagonist HOE140 for 30 min and then stimulated with 10 nM LDBK. **P < 0.01 respect to cells stimulated only with LDBK. Images are representative of three independent experiments and data correspond to the mean ± SEM (n = 3). VEGF-R2: vascular endothelial growth factor receptor-2; bp: base pair.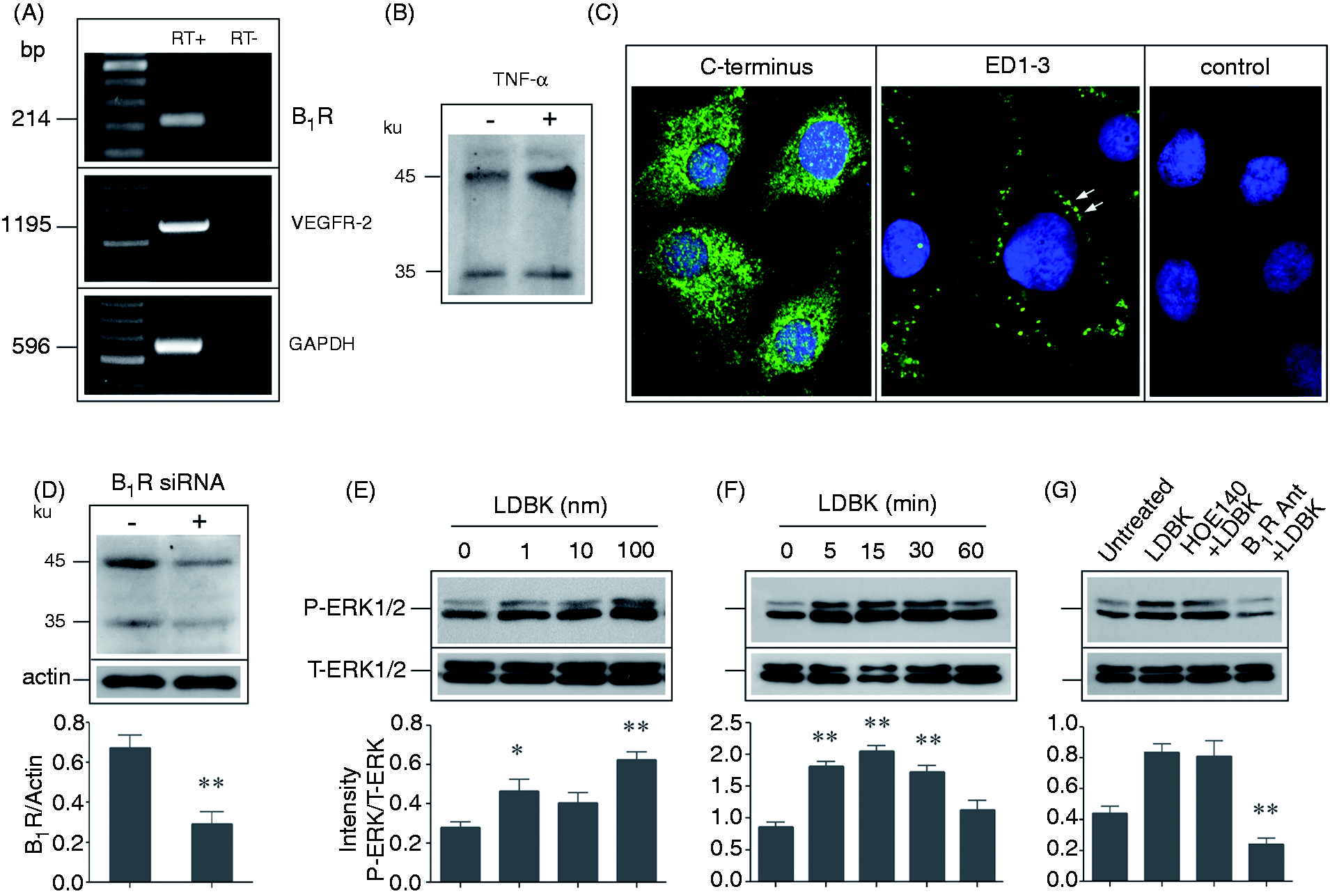
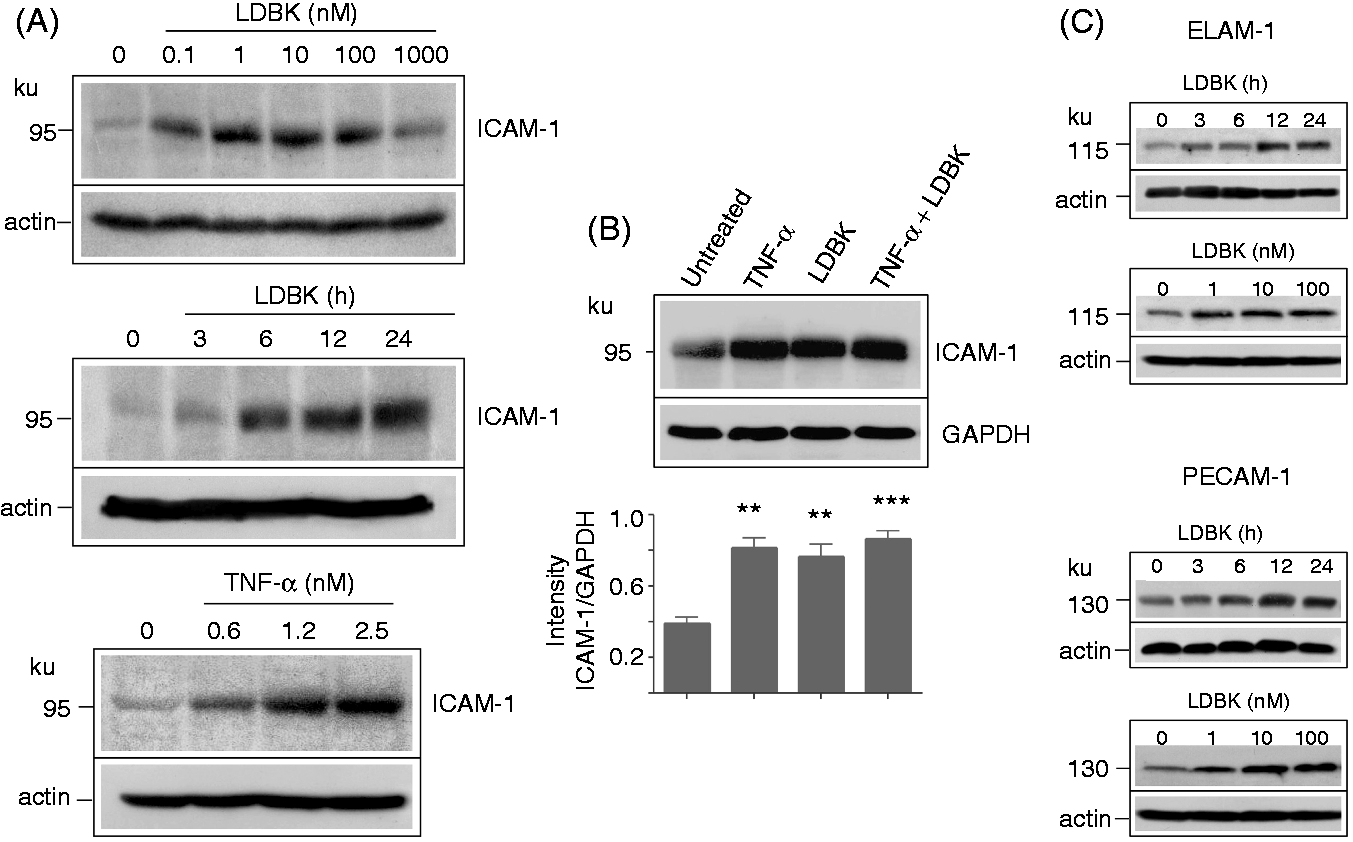
Once the presence of B1R was established on endothelial cells they were stimulated with LDBK, and dose- and time-dependency determined. RT-PCR showed that stimulation of B1R with 1–100 nM LDBK resulted in up-regulation of ICAM-1 transcripts, an effect that was observed after 3–6 h of stimulation (Figure 6A). In control experiments, this result was confirmed using TNF-α, a cytokine that is known to up-regulate ICAM-1 after 3 or 6 h of stimulation (Figure 6C). Quantitative PCR showed that stimulation of endothelial cells with 100 nM LDBK for 3 h produces a significant up-regulation of ICAM-1 mRNA when compared with basal levels in untreated cells (Figure 6B). Using an identical experimental approach, we found that ICAM-1 protein, assessed by Western blots of endothelial cell homogenates, was also up-regulated as a consequence of B1R stimulation. Furthermore, it was observed that levels of ICAM-1 protein increased significantly after endothelial cells were stimulated with 0.1–100 nM LDBK for a period that ranged between 6 and 24 h (Figure 7A). This effect was confirmed using TNF-α as a positive control (Figure 7A). Furthermore, stimulation of endothelial cells with 10 or 100 nM LDBK produced an up-regulation of ICAM-1 that was comparable with that produced by 1 nM TNF-α (Figure 7B). Interestingly, B1R stimulation not only up-regulated ICAM-1 protein, but also other intercellular adhesion molecules such as ELAM-1 and PECAM-1; like ICAM-1, levels of both molecules increased when concentrations of LDBK as low as 1 nM were used (Figure 7C). Incubation of cells with the solvents used to suspend the B1R agonist (0.9% NaCl), did not up-regulate ICAM-1 or other intercellular adhesion molecules (not shown).
B1R stimulation increases the expression of ICAM-1. Monolayers of EA.hy926 endothelial cells were stimulated with different concentrations of LDBK for variable periods. The total RNA was extracted and the cDNAs were amplified by conventional (A) or quantitative PCR (B). Positive controls were performed using endothelial cells stimulated with 1 nM TNF-α (C). Data represent the mean ± SEM of three independent experiments (n = 3). *P < 0.05 between LDBK-stimulated and untreated cells. bp: base pair. B1R stimulation increases the protein levels of ICAM-1, ELAM-1 and PECAM-1. (A) Endothelial cells were stimulated with various concentrations of LDBK for 12 h or with 100 nM LDBK for variable periods of time. Positive controls were performed stimulating another group of cells with different concentrations of TNF-α. (B) Representative experiment in which endothelial cells were stimulated with 1 nM TNF-α, 100 nM LDBK or both for 12 h. Proteins were separated by SDS-PAGE, transferred onto Immobilon P membranes and immunoprinted for ICAM-1 (A, B), ELAM-1 (C) and PECAM-1 (C). Western blots are representative of three independent experiments (n = 3) and data represent the mean ± SEM.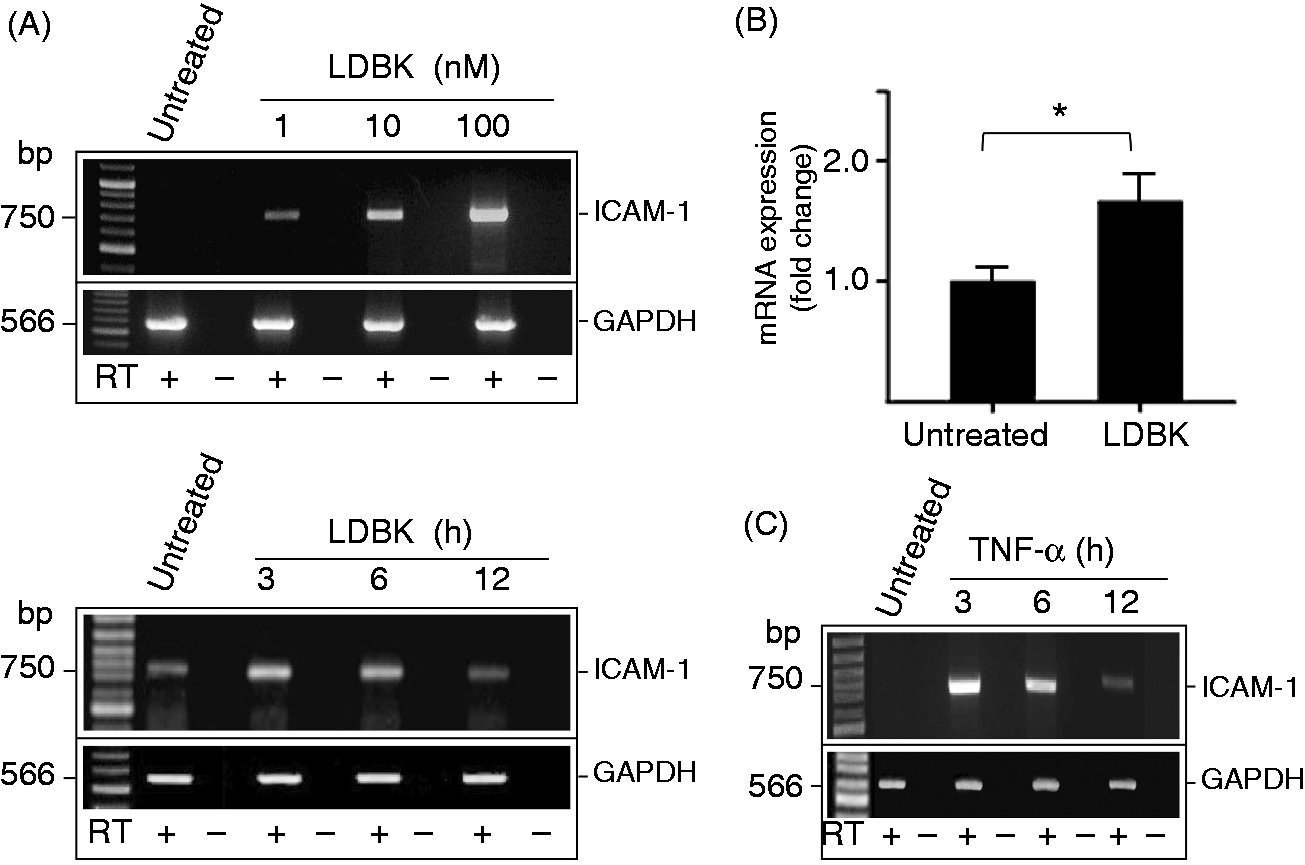
The stimulatory effect of LDBK on the expression of ICAM-1 was almost entirely blocked when endothelial cells, which had been transfected previously with a commercial B1R siRNA for 24 and 48 h, were stimulated with 10 nM LDBK under comparable conditions (Figure 8A). Treatment of cells with this siRNA reduced the amount of B1R protein in membrane fractions when analyzed by Western blots (Figure 5D). Specificity of the siRNA had previously been assessed in parallel experiments in which the levels of B1R transcripts were determined by RT-PCR after 24 and 48 h of transfection of the siRNA.
27
Additional control experiments, in which endothelial cells were pretreated with an excess amount of a B1R antagonist (1 µM) prior to stimulation with 10 nM LDBK, indicated that LDBK had increased ICAM-1 levels by activating kinin B1R (Figure 8B).
ICAM-1 expression is blocked by a B1R antagonist or after silencing of the B1R with a siRNA. (A) Transfected and non-transfected endothelial cells were stimulated with 10 nM LDBK for 24 h. (B) Cells were directly stimulated with 10 nM LDBK or after pre-incubation for 30 min with the B1R antagonist (B1R Ant) des[Arg9]Leu8-bradykinin (1 µM). Data represent the mean ± SEM of three independent experiments (n = 3). *P < 0.05 between LDBK stimulated and cells pretreated with a B1R antagonist or transfected with a B1R siRNA.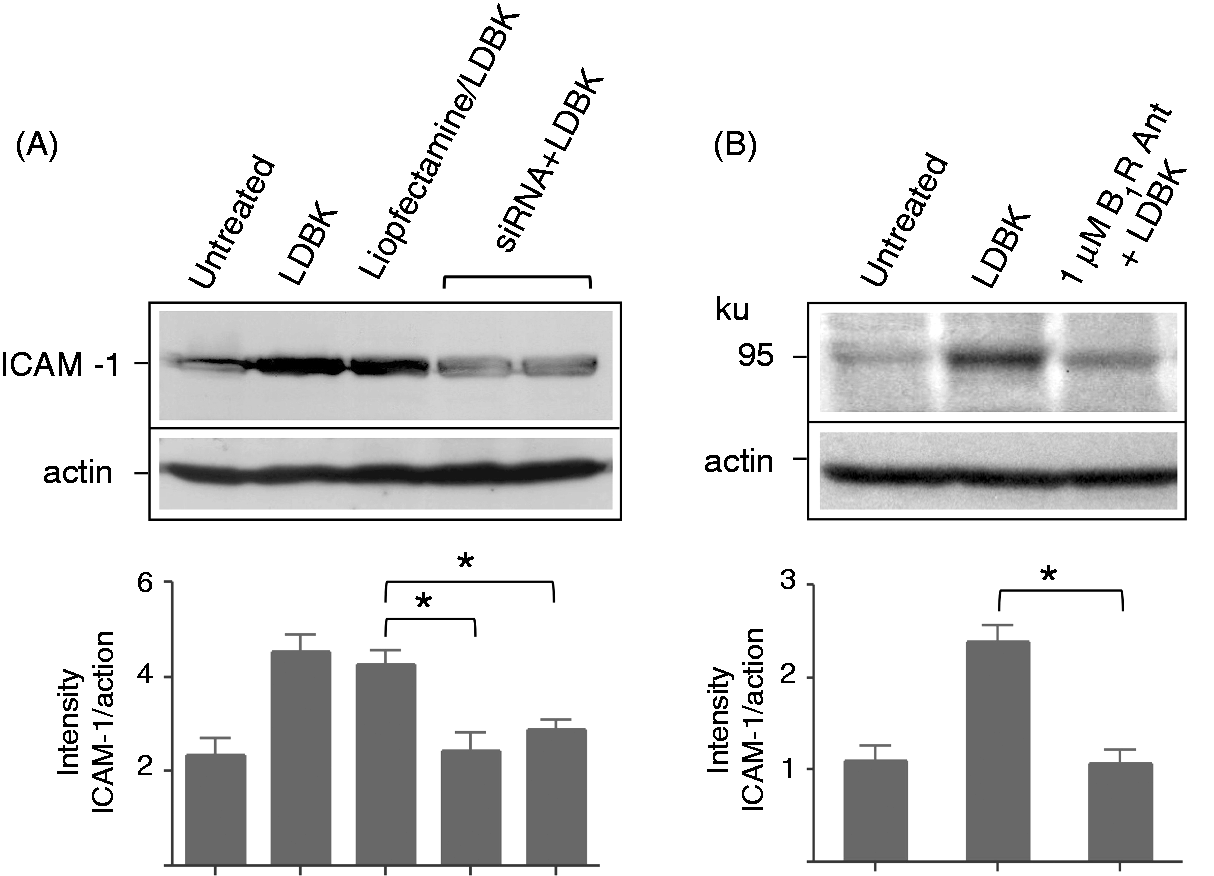
Immunolabeling experiments indicated that ICAM-1 levels were lower in non-stimulated endothelial cells when compared with those stimulated either with LDBK or TNF-α (Figure 9). Confocal optical sectioning of the endothelial cells every 0.4 µm from top to bottom revealed that immunolabeled ICAM-1 in stimulated cells was concentrated on filopodia exposed on the luminal side as evident after three-dimensional (3D) reconstruction of the endothelial luminal surface (Figure 9B, C).
Visualization of immunoreactive ICAM-1 in endothelial cells stimulated with LDBK. (Upper row) Cells were synchronized for 12 h in the absence of FBS prior to stimulation with TNF-α or LDBK. Next, they were fixed with absolute methanol and immunostained with an anti-ICAM-1 Ab followed by Alexa-488 labeled Igs. Nuclei were stained with DAPI and optical sections were taken every 0.4 µm from top to bottom. (Bottom row) High magnifications of untreated (A), LDBK-stimulated endothelial cells (B, C) and positive control using TNF-α as stimulus (D). (C) A 3D reconstruction of the endothelial immunoreactive luminal surface. Arrows point out luminal filopodia. Images are representative of three independent experiments.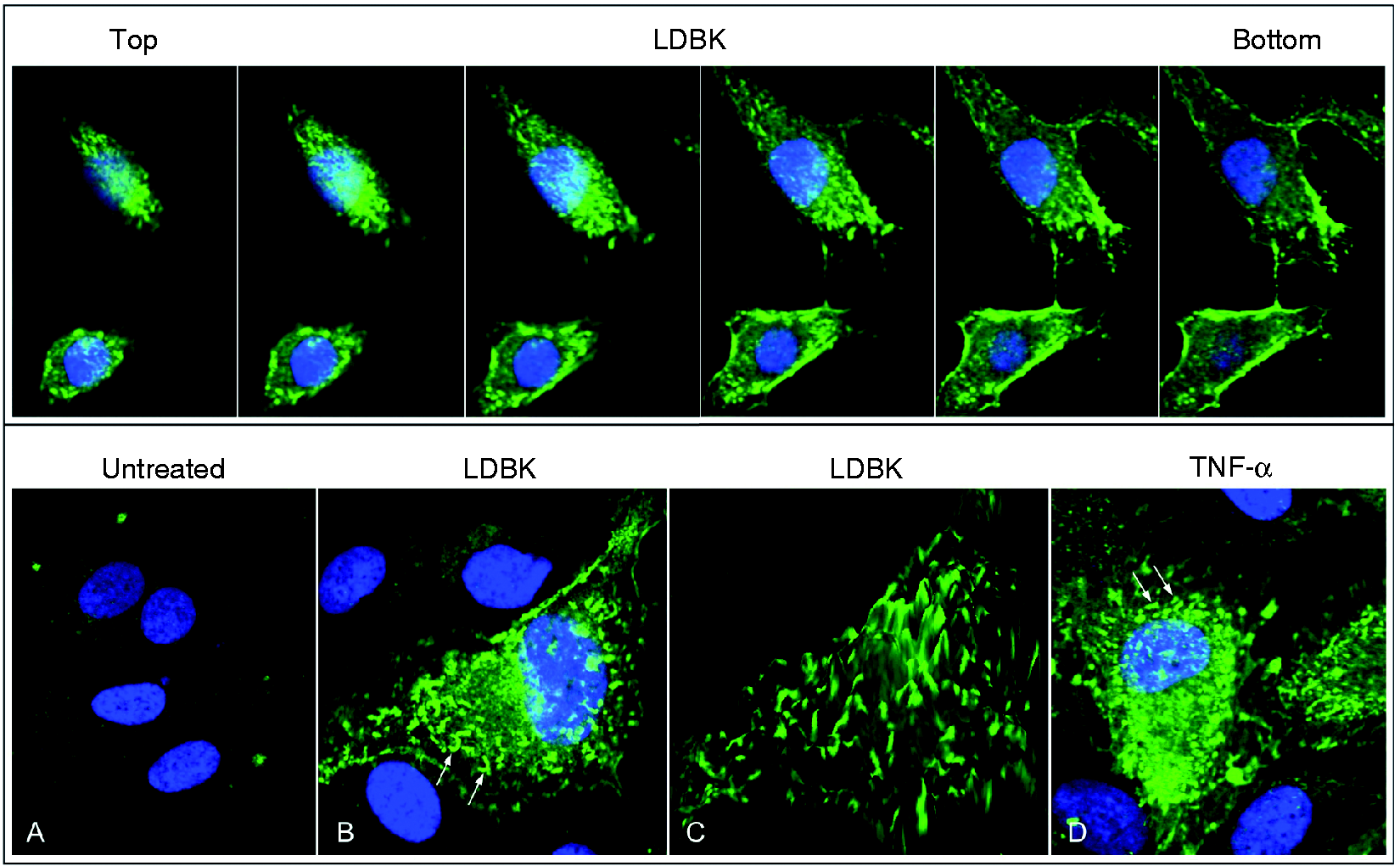
Stimulation of B1R increases adhesion between neutrophils and endothelial cells
As the experimental data indicated that stimulation of B1R by LDBK increases levels of neutrophil integrins and endothelial cell intercellular adhesion molecules, we designed experiments to examine the question whether co-culture of these two cell types would result in interactions between their molecular families. Stimulation of a post-confluent endothelial cell monolayer with a range of concentrations of LDBK for 8 h produced firm adhesion of neutrophils (previously activated with 1 nM TNF-α for 30 min; Figure 10A–C). When the endothelial monolayer was pretreated with a B1R antagonist (10 µM), prior to stimulation with 100 nM LDBK or assay performed in the presence of a neutralizing anti-ICAM-1 Ab, adhesion between monolayer and neutrophils was reduced considerably (Figure 10C). Further experiments confirmed that neutrophils, when stimulated with 100 nM LDBK for 30 min, become tightly attached to the endothelial monolayer (pre-activated with 1 nM TNF-α for 8 h). Similarly, pretreatment of neutrophils with either a B1R antagonist or a neutralizing Ab directed to Mac-1 during the assay decreased attachment between the two cells (Figure 10A, D).
Neutrophil-endothelial cell adhesion assay. (A) Representative images of experiments (n = 5) in which endothelial cells and neutrophils were stimulated with TNF-α, TNF-α/LDBK or LDBK/TNF-α, respectively, or neutrophils were pretreated for 30 min with a B1R antagonist prior stimulation with LDBK and incubation with endothelial cells previously activated with TNF-α. (B) The monolayer was stimulated with different concentrations of LDBK and neutrophils were stimulated with TNF-α. (C) Human neutrophils were stimulated with TNF-α, whereas the post-confluent endothelial monolayer was stimulated with 100 nM LDBK. Controls included stimulation of neutrophils and endothelial cells with TNF-α (TNF-α/TNF-α), pretreatment of the monolayer with a B1R antagonist (10 µM B1R Ant) prior stimulation by LDBK or presence of a neutralizing anti-ICAM-1 Ab during the assay. (D) The endothelial cell monolayer was stimulated with TNF-α, whereas neutrophils were stimulated with 100 nM LDBK. Controls included stimulation of neutrophils and endothelial cells with TNF-α, pretreatment of neutrophils with a B1R antagonist (10 µM) prior stimulation by LDBK or presence of a neutralizing anti-Mac-1 Ab during the assay. Data represent the mean ± SEM of five independent experiments (n = 5). *P < 0.05; **P < 0.01.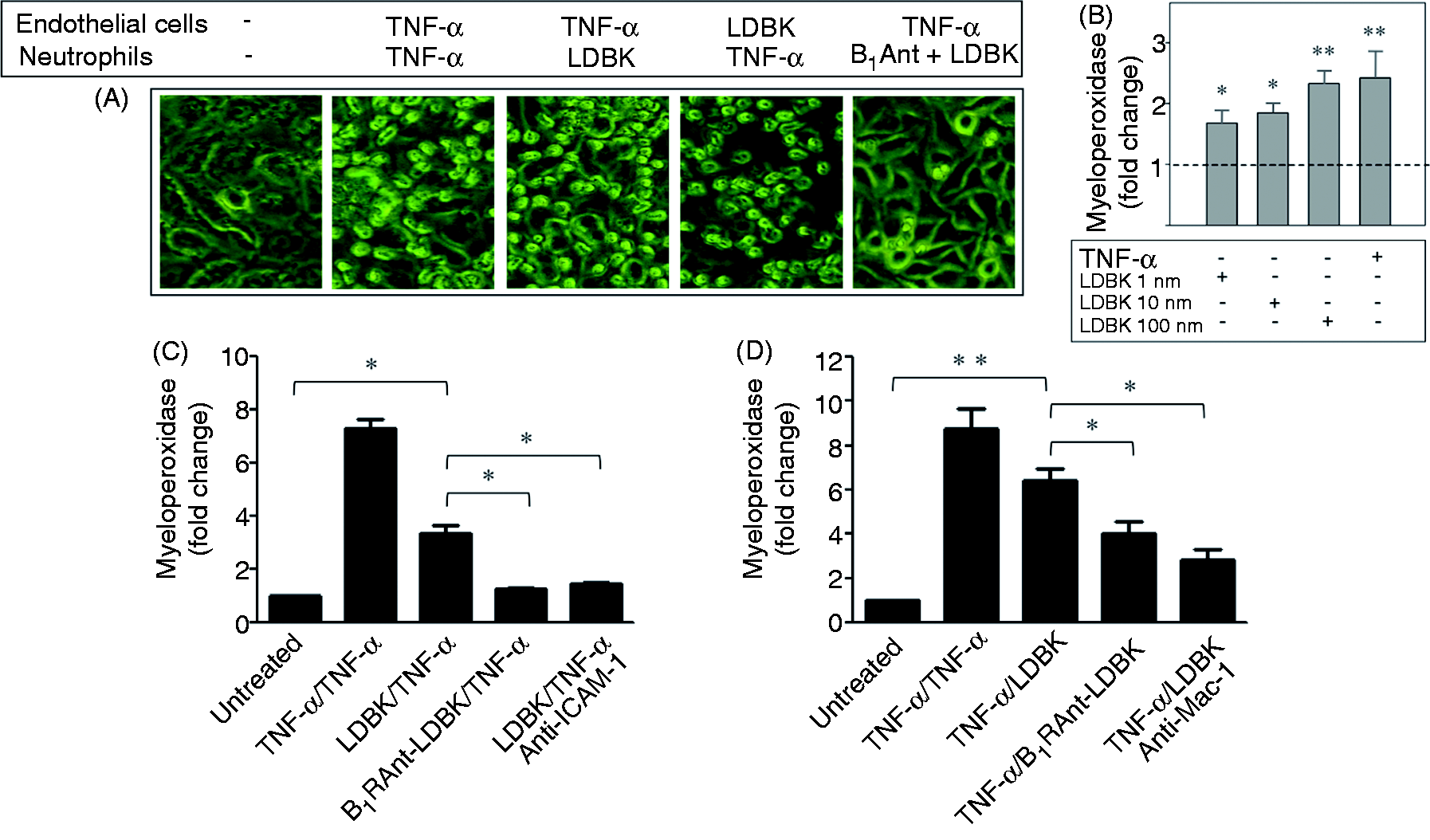
Discussion
Early studies have indicated that chemotactic factors prompt leukocytes and endothelial cells to attach to one another by activating specific families of adhesion molecules, a key step for the subsequent recruitment of leukocytes into inflamed tissue sites. 31 Kinins are pro-inflammatory peptides considered to play a major role in acute inflammation as they accumulate at the sites of injury and mimic the cardinal signs of inflammation.6,7 Because kinins stimulate neutrophil chemotaxis, they may also favor leukocyte trafficking across the endothelial wall by using transcellular and/or paracellular transmigration.12,21,32,33 As the question of whether kinin receptor agonists are able to modify the expression patterns of endothelial adhesion molecules and/or neutrophil integrins has not been comprehensively addressed, this study was designed to examine this pertinent question. We demonstrate herein that levels of the integrins LFA-1 and Mac-1 are increased when human neutrophils are stimulated with LDBK. Furthermore, the increased levels were reduced when the cells were pre-incubated with a B1R antagonist prior to stimulation with the agonist. LFA-1 has been shown to regulate the rolling velocity, adhesion and migration of leukocytes, whereas Mac-1—by interacting with ICAM-1—its counter-receptor on endothelial cells, induces firm capture of neutrophils to the endothelium. 34 The effect of B1R activation on the expression of Mac-1 may go beyond adherence, as some authors have reported that β2-integrins may be involved in (i) the binding of C3bi coated particles to phagocytes, (ii) platelet–neutrophil aggregation and (iii) the binding of neutrophils to endothelial cells. Mac-1 also binds fibrinogen, the activation of which results in the stimulation of phagocytosis, the respiratory burst, granule enzyme release, adhesion, spreading and cell polarization.21,34 Importance of the integrins is evident in disorders such as vasculitis in which β2-integrins and ICAM-1, together with α4β1 integrins and VCAM-1, mediate the firm adhesion of neutrophils and guide them through transendothelial migration. 35 In experiments using flow cytometry and Western blotting, we showed that incubation of whole blood with the B1R agonist increased the exposure of Mac-1 on the neutrophil surface after 15 min of incubation. Interestingly, prolonged incubation of whole blood or isolated neutrophils with LDBK resulted in a marked increase in Mac-1. In the study by Shima et al. 36 on the analysis of the degradation products of bradykinin in human plasma by reverse-phase HPLC, in the time course of degradation of des[Arg9]bradykinin, this analogue of LDBK was maximally stable at 90 min. Therefore, in the experiment in which the stimulatory effect of LDBK on Mac-1 was determined over an incubation period of 15 min, LDBK was considered to be both stable and functional.
Although the increase on the cell surface LFA-1 of LDBK-stimulated neutrophils was small, it was augmented significantly when analyzed by Western blots, suggesting that the additionally observed increase may correspond to intracellular stored LFA-1. Increase of the neutrophil integrins also occurred at low concentrations of the kinin B1R agonist (10−9 and 10−8 M), making it physiologically relevant to the inflammatory milieu. Cycloheximide, a protein synthesis inhibitor reduced the increment of Mac-1 or LFA-1 produced by LDBK suggesting that de novo synthesis of integrins may occur also. Similarly, pretreatment of neutrophils with brefeldin A for 60 min produced an accumulation of integrins in the stimulated neutrophils. Our results support those reported by Zhou et al., 37 who determined that TLR4-activated CD11b expression was cycloheximide-sensitive within 3 h of challenge with LPS. Interestingly, the majority of known stimuli that favor expression of these types of adhesion molecules belong to the family of G protein-coupled receptors that includes kinin B1R. Nevertheless, it is necessary to show increased RNA expression to convincingly conclude that these integrins are newly expressed following B1R stimulation.
Integrins exposed on the neutrophil surface have to make contact with the intercellular adhesion molecules expressed on endothelial cells before they leave the microcirculation and migrate to the inflamed site. It is well known that upon stimulation by inflammatory cytokines, such as TNF-α, IL-1β and IFN-γ, endotoxins and cellular stress, among others, intercellular adhesion molecules are highly up-regulated to support the migration of leukocytes to sites of infection or inflammation.38–40 Our results indicate that expression of both ICAM-1 mRNA and protein is up-regulated in endothelial cells stimulated by LDBK, similarly to that produced by TNF-α. Interestingly, B1R stimulation also increased the levels of PECAM-1 and ELAM-1 (E-selectin) proteins, suggesting that B1R may be involved not only in the functional adhesion of neutrophils to endothelial cells, but also in the rolling phase prior to adhesion. Other inflammatory mediators, such as substance P, promote increased neutrophil transendothelial migration through ICAM-1 up-regulation. 41 Multiple signaling pathways, such as protein kinase C, Ca2+, Rho kinase, ERK1/2, p38 and JNK MAPK, NF-κB and Src are believed also to regulate expression, activation and clustering of adhesion molecules on the endothelial cell surface.42,43 Our preliminary results appear to indicate that the B1R utilizes signaling pathways, such as protein kinase C, PI3K and MAPKs, to regulate the expression of intercellular adhesion molecules on endothelial cells as pretreatment with inhibitors such as GF109203X, LY294002 or PD98059 prior to stimulation with LDBK reduced the expression of ICAM-1 and PECAM-1 (data not shown). In this respect, up-regulation of ICAM-1 by LPS is mediated by p38 MAPK at the level of gene transcription. 44 Similarly, TNF-α increases mRNA levels and enhances expression of ICAM-1 on the endothelial cell surface by a pathway that seems to involve phospholipase C and protein kinase C. 45
It has been shown recently that firm adhesion of neutrophils to endothelial cells via ICAM-1 promotes their transmigration together with PECAM-1. Liu et al. 43 reported a functional relationship between ICAM-1 activation and PECAM-1 binding activity based on the fact that an increase in PECAM-1 binding is reduced in human umbilical endothelial cells expressing a phosphorylation-defective ICAM-1 mutant. As PECAM-1 is also up-regulated by LDBK, this observation suggests that B1R may have an important role in vivo, favoring the diapedesis of neutrophils. 43
Upon binding to β2 integrins, ICAM-1 is clustered on the endothelial cell membrane and triggers intracellular signaling that results in a ring of dynamic membrane protrusions named filopodia, a process involving rearrangement of the actin cytoskeleton, local increase in F-actin content and participation of small GTPases.46,47 In our study, using confocal microscopy, we have shown that LDBK induces filopodia formation on the surface of endothelial cells that are similar to those formed as a result of stimulation with TNF-α, and are absent on unstimulated cells.
The next question we addressed was whether ICAM-1 and integrins, expressed in endothelial cells and neutrophils respectively, were active functionally. To answer this issue, we used an in vitro adhesion assay in which post-confluent monolayers of endothelial cells, stimulated with LDBK to up-regulate ICAM-1, were able to adherē to more neutrophils than unstimulated monolayers. In fact, dose-dependent adhesion of neutrophils to the monolayer stimulated with different concentrations of LDBK was observed. Similarly, LDBK-stimulated neutrophils adhered to the endothelial monolayer in amounts comparable with those produced by TNF-α. The use of neutralizing Abs to ICAM-1 and Mac-1 in the assay diminished adhesion between endothelial cells and neutrophils, whereas a B1R antagonist confirmed that adhesion was dependent on B1R activation. Considering that LDBK-stimulated neutrophils did not show significant levels of LFA-1 on the cell surface, it is reasonable to assume that adhesion to the endothelial monolayer is primarily through an interaction between ICAM-1 and Mac-1 rather than LFA-1.
Up-regulation of ICAM-1 and Mac-1 by LDBK may go beyond adhesion and transendothelial migration as it has been shown that adhesion of neutrophils to endothelial cells through Mac-1/ICAM-1 interaction produces changes in transepithelial electrical resistance which promotes permeability to albumin, an effect that can be diminished by anti-ICAM-1 Abs. 48 This finding is supported by the early studies of Wedmore and Williams, 49 who demonstrated that tissue edema after administration of an inflammatory stimulus was dependent upon neutrophils, as well as endothelial cells. They showed that application of an inflammatory mediator in animals depleted of circulating neutrophils abolished tissue edema. The use of an anti-CD18 Ab produced identical results, 50 suggesting that firm adhesion of neutrophils to endothelial cells is an important and early event during an increase in vascular permeability. Gautam et al.51,52 provided evidence that adhesion or ligation of CD11b/CD18 induces the release of cationic proteins, azurocidin, elastase, cathepsin G and proteinase-3—an event that increases permeability. They reported that neutrophil-derived azurocidin was responsible for the increase in vascular permeability produced after adhesion. We, and others, have shown that human neutrophils, as well as endothelial cells, possess on their surface an assembly of essential components for the contact activation system (solid phase).10,53 Based on this observation and on the long-known effects of kinins in the microcirculation,15,54 we had hypothesized 10 that a discrete and circumscribed formation of kinin peptides from kininogens by plasma kallikrein and/or by release of neutrophil-borne KLK111,13 would permit the exudation of plasma content into the inflamed tissue. The results obtained in the current study provide additional evidence for our hypotheses that kinins facilitate the migration of neutrophils into the interstitial tissue space surrounding the site of injury.
In summary, we have shown that (i) endothelial cells exposed to LDBK can increase adhesiveness to neutrophils and vice versa; (ii) increased adhesiveness is due to an up-regulation of cell surface ICAM-1/PECAM-1 in endothelial cells and Mac-1 on the neutrophil cell membrane because neutralizing Abs to ICAM-1 or Mac-1 diminished adhesion between both cells; and, finally, (iii) enhanced adhesiveness is produced as a result of B1R activation as a siRNA and a B1R antagonist blunted the expression of ICAM-1 in endothelial cells, and the B1R antagonist blocked the up-regulation of Mac-1 and LFA-1 in neutrophils and adhesion in an in vitro adhesion assay. We also found that LDBK increases the expression of ELAM-1, a molecule involved in the rolling phase of neutrophils that occurs prior to firm adhesion and diapedesis. These findings position B1R as a novel modulator of neutrophil adhesion and suggest that its inhibition may attain major importance during acute inflammation or during acute episodes of chronic inflammatory disorders that require recruitment of a large population of neutrophils to the inflamed site.
Footnotes
Funding
This work was supported by the Fondo Nacional de Investigacion Cientifica y Tecnologica (FONDECYT, Chile) [grant number 1110464].
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
Acknowledgements
We wish to thank Mr. Manuel Barria for his technical support, and Merck & Co, West Point, PA, USA and Prof. Werner Müller-Esterl of Goethe University, Frankfurt, Germany for their kind gift of anti-B1 receptor Abs.