
Abstract
Respiratory burst function of neutrophils is thought to play a pivotal role in the development of pathologies such as indirect (extra-pulmonary) acute lung injury (iALI), as well as sepsis. The current study was conducted to determine the effect of an HIV transactivator of transcription (TAT)-fusion protein containing a soluble N-ethylmaleimide-sensitive factor attachment protein receptor domain from synaptosome-associated protein-23 (SNAP-23) on the shock/sepsis- and sepsis-enhanced neutrophil burst capacity using the clinical relevant two-hit iALI mouse model and the classical cecal ligation and puncture (CLP) septic model. TAT-SNAP-23 significantly decreased the blood neutrophil respiratory burst in vitro, and also in vivo in CLP and hemorrhaged mice. We found that the neutrophil influx to the lung tissue, as measured by myeloperoxidase levels and neutrophil-specific esterase+ cells, was also decreased in the TAT-SNAP-23-treated group. Consistent with this, treatment of TAT-SNAP-23 significantly reduced the disruption of lung tissue architecture and protein concentration of bronchoalveolar lavage fluid in iALI mice compared with vehicle-treated iALI mice. In addition, although TAT-SNAP-23 did not alter the extent of local cytokine/chemokine expression, the in vitro migration capacity of neutrophils was blunted from septic and hemorrhagic mice. These data support our hypothesis that TAT-SNAP-23 reduces neutrophil dysfunction in iALI and sepsis by inhibiting neutrophil respiratory burst.
Introduction
Sepsis is a progressive, inflammatory response to overwhelming infection associated with tissue hypoperfusion and multi-organ dysfunction.1,2 Despite the development of novel treatments such as early appropriate antibiotic therapy, and early goal-directed therapy, severe sepsis remains a common and frequently fatal condition with high associated costs and a mortality rate approaching 50% in the case of septic shock.1–3 Acute lung injury (ALI) and its most severe form, acute respiratory distress syndrome (ARDS), are characterized by increased capillary and alveolar permeability, hypoxemia, decreased lung compliance and diffuse bilateral pulmonary infiltrates. 4 At present, therapeutic interventions to treat ALI or ARDS remain limited. Thus far, no real pathophysiologic-driven therapeutic interventions have been available.5,6 Based on pathophysiologically-oriented views, ALI and ARDS can be of pulmonary (direct) or extrapulmonary (indirect) origin. 7 Indirect ALI (iALI) accounts for approximately 20% of all ALI cases; meanwhile, sepsis is the most commonly encountered condition underlying the development of ALI, with severe sepsis accounting for 33% of indirect ALI cases. 8 Several studies have indicated that direct and indirect ALI are truly different.9–12 With the failure of many supportive therapies in ARDSNet,5,6 novel therapeutic strategies focusing on etiology and mechanism are still needed.
Neutrophils comprise one of the major cellular components of the innate immune system. They are thought to contribute directly to antimicrobial killing via expression of a range of antimicrobial peptides, proteases and oxidants, which are proposed to have an important role in sepsis and inducing iALI.13–16 There is compelling evidence indicating that neutrophil function during severe sepsis and iALI is substantially dysregulated, resulting in impaired directed migration of neutrophils to infectious foci and inadequate antimicrobial responses.13,14 The ability of neutrophils to destroy bacteria or other pathogens is dependent upon the ability to mount an effective respiratory burst response.17–19 Respiratory burst function resulting in the release of reactive oxygen species (ROS) such as superoxide anion (O2−) from neutrophils is one of the mechanisms of the innate immune response to a microbial challenge. Maladaptive control of this mechanism is thought to play a pivotal role in the development of iALI/ARDS, as well as sepsis,17–19 and both are reported to be the most common forms of organ dysfunction seen in trauma/surgical intensive care unit patients. Using our clinically relevant hemorrhagic shock [hemorrhage (Hem); a ‘priming’ insult] followed by polymicrobial septic challenge [cecal ligation and puncture (CLP); ‘triggering’ event] (a double-hit model that has been demonstrated to induce iALI) animal model, we reported previously that neutrophils isolated from donor animals that had undergone hemorrhagic shock were capable of inducing iALI when adoptively transferred into naive (neutrophil-depleted) animals that were subsequently subjected to a septic challenge.19–21 In this setting, in vivo neutrophil ‘priming’ by Hem was not only associated with an increase in respiratory burst capacity ex vivo, but also with a decrease in neutrophil apoptosis,19–21 a finding confirmed by others in experimental iALI. 22 We further found that these primed neutrophils were recruited into the lungs to mediate the development of ALI upon exposure to the second hit, while the severity of iALI was reduced by neutrophil depletion 23 or the blockade of CXC receptor 2 signaling. 24 Although a role for neutrophil respiratory burst capacity in sepsis and iALI is well established, strategies to control neutrophil activation to prevent tissue and organ injury have not followed.
To kill microbial organisms more effectively, the neutrophil respiratory burst, based upon the assembly and activation of NADPH oxidase, which then catalyses the univalent reduction of molecular oxygen (O2) to O2−, is augmented by a variety of biological agents through a process termed ‘priming’.14,25 The respiratory burst can be primed by exocytosis of neutrophil granule subsets.26,27 Soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) proteins play a central role in intracellular membrane trafficking by mediating fusion of membranes from different cellular compartments.28–30 It has been suggested that the selective SNARE proteins in different granule membranes could have their independent mobilization of the different granule populations present in neutrophils during activation.31,32 Studies have also shown that synaptosome-associated protein-23 (SNAP-23), one of the SNARE proteins that has been identified on neutrophil granules and plasma membranes, plays a role in neutrophil granule exocytosis. 33 Uriarte et al. 33 recently developed an HIV transactivator of transcription (TAT)-fusion protein containing a SNARE domain from SNAP-23 (TAT-SNAP-23). They demonstrated TAT-SNAP-23’s ability to inhibit TNF-α and platelet-activating factor-induced priming of the normal human neutrophil respiratory burst by up to 80% in vitro by blocking exocytosis selectively. Further, they confirmed that neutrophil granule exocytosis contributing to phagocytosis-induced respiratory burst played a critical role in priming of the respiratory burst by increasing expression of membrane components of NADPH oxidase. 33 They recently showed that i.v. administration of TAT-SNAP-23 resulted in amelioration of injury in a rat model of direct ALI (via immune complex deposition) by inhibiting neutrophil exocytosis. 34 However, to our knowledge, the action of TAT-SNAP-23 on neutrophil respiratory burst in a murine model of sepsis or iALI has not been elucidated. Therefore, it is quite reasonable to hypothesize that TAT-SNAP-23 could have a role in regulating neutrophil respiratory burst induced by murine models of sepsis and Hem-induced priming for iALI. The goal of the present study was to identify TAT-SNAP-23 as an inhibitor of neutrophil respiratory burst in vivo using our two-hit model of iALI or CLP alone.
Materials and methods
Mice
Male C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice aged 8–10 wk were used for experiments. Experiments were performed in accordance with National Institutes of Health guidelines and with approval from the Animal Use Committee of Rhode Island Hospital.
Reagents
Keratinocyte-derived chemokine (KC) and macrophage inflammatory protein-2 (MIP-2) Abs for ELISA assays were purchased from R&D Systems (Minneapolis, MN, USA). Mouse IL-6, IL-10, monocyte chemoattractant protein-1 (MCP-1) and TNF-α ELISA kits were purchased from BD Bioscience (San Diego, CA, USA). The TAT-SNAP-23 fusion protein was provided by Uriarte et al.33,34 All other chemicals were of analytical reagent grade and purchased from Sigma Chemical (St Louis, MO, USA).
Rodent models
Hem
The non-lethal fix-pressure Hem model used for these experiments has been previously described.19–21,23,24 In brief, mice were anesthetized with isoflurane, restrained in supine position, and catheters were inserted into both femoral arteries. Anesthesia was discontinued, and blood pressure was continuously monitored through one catheter attached to a blood pressure analyzer (MicroMed, Louisville, KY, USA). When fully awake, as determined by a mean blood pressure of ∼ 95 mmHg, the mice were bled over a 5–10-min period to a mean blood pressure of 35 mmHg (± 5 mmHg) and kept stable for 90 min. Immediately after Hem, mice were resuscitated with Ringer’s lactate at four times drawn blood volume. After resuscitation, arteries were ligated, catheters removed, and catheter sites bathed with lidocaine and sutured closed. Sham mice were anesthetized, restrained in a supine position for the same duration, and blood vessels were ligated, not bled.19,21,23,24
Polymicrobial sepsis/CLP
Mice were anesthetized with isoflurane and restrained in supine position. A 1-cm midline incision was made; the cecum was ligated with 5-0 silk thread and punctured twice with a 22-gauge needle. The cecum was then replaced, and the incision was closed. Mice were resuscitated with 1 ml Ringer’s lactate subcutaneously and returned to their cages. Sham mice were anesthetized, restrained in a supine position for the same duration and the cecum was exposed, but not ligated nor punctured.19–21,23,24
iALI (Hem/sepsis model)
Mice subjected to Hem procedures 24 h later, sepsis was induced as a secondary challenge via CLP as previously described.19–21,23,24 It should be noted that in either CLP alone or the combined insults of Hem followed by CLP all animals were euthanized for study prior to the development of mortality that often accompanies these models.
Neutrophil isolation
Neutrophil isolation from individual mouse blood was performed as previously described.19,20 In summary, whole blood was collected from naive, sham CLP, CLP, Hem alone or iALI mice via cardiac puncture into a heparin-rinsed syringe. An equal volume of 3% Dextran in PBS was added to the heparinized blood, mixed vigorously and left to stand at room temperature for 45 min. The top leukocyte rich layer was removed, transferred to a 15-ml tube, PBS added to 15 ml in total and centrifuged for 10 min at 400 g (at 4℃). Cells were re-suspended in 3 ml PBS with 0.1% BSA and layered over a discontinuous density percoll gradient (2 ml of 1.097 density in the bottom layer carefully overlaid with 3 ml of 1.077 density) and centrifuged for 1 h at 500 g (at 25℃). Neutrophils were removed from lower interface, transferred to a 15-ml tube and filled up with PBS to 15 ml, centrifuged for 15 min at 805 g (at 4℃). The number of viable cells per sample was determined using Trypan blue exclusion.
Respiratory burst assay
In vitro
Isolated neutrophils (3 × 105 cells) from naive or sham mice were pretreated with increasing concentration of TAT-SNAP-23 (0.2–0.6 µg/assay) in the presence or absence of TNF-α, as a priming agent (2 ng/ml, at 37℃ for 10 min), followed by phorbol 12-myristate 13-acetate (PMA; 20 nM). Superoxide release was assessed by measuring the superoxide dismutase-inhibitable reduction of ferricytochrome C. After reading at duel wavelength 550, 630 (reference wavelength) at 37℃, data were analyzed using the following formula: Respiratory burst = 13.258 × (OD at X min – OD at 0 min). 19
In vivo
For the in vivo studies, Sham and CLP mice were given 6 or 12 µg of TAT-SNAP-23 in 200 µl Hank’s balanced salt solution (HBSS)/mouse by i.v. tail injection right after surgery. Hem mice were given 12 or 24 µg of TAT-SNAP-23 in 200 µl HBSS/mouse by i.v. infusion during resuscitation period. The dose of TAT-SNAP-23 used for these experiments was determined based on the results of TAT-SNAP-23 dose study in vitro. Blood was collected and neutrophils were isolated at various times after surgery, and the respiratory burst activity was assessed as described in the section ‘In vitro’.
Neutrophil chemotaxis assay
The modified Boyden chamber/48-well micro-chemotaxis assay was performed as per the manufacturer’s protocol (Neuro Probe, Gaithersburg, MD, USA). Briefly, 25 µl of formyl-methionyl-leucyl-phenylalanine (fMLP) (stock 5 nM) or media control (DMEM) was pipetted into the 48 wells in the bottom chamber. Isolated neutrophils from CLP and hemorrhaged mice were pipetted into the top wells (50 µl media containing 1 × 104 cells/well). The chamber was incubated for 60 min at 37℃. Following incubation, the chamber was inverted and the filter removed. Non-migrated cells were wiped off from the cell solution side of the membrane and the migrated cells on the membrane were stained with modified Giemsa stain (Sigma-Aldrich, St Louis, MO, USA) and counted at 100 × magnification on at least five random fields. Results were expressed as number of neutrophils per field. The number of migrating cells present in the containing media alone condition were used as controls for random migration. 21
Lung myeloperoxidase activity
As an assessment of neutrophil influx, myeloperoxidase (MPO) activity was measured according to established protocols.20,21 Lung tissue was homogenized in 0.3 ml 50 mM potassium phosphate buffer (0.1 M of KH2PO4 and 0.1 M of K2HPO4), centrifuged at 500 g (at 4℃) for 5 min; then, the supernatant was collected and centrifuged at 20,000 g (at 4℃) for 60 min. The pellet was then re-suspended in 0.3 ml sonicate buffer [50 mM potassium phosphate buffer (0.1 M of KH2PO4, 0.1 M of K2HPO4 and 0.5% of hexadecyltrimethyl-ammonium bromide)] for 30 s. After three freeze–thaw cycles, lysate was centrifuged 12,000 g (at 4℃) for 10 min. Supernatants were then assayed for MPO activity at a 1:20 dilution in the reaction buffer [50 mM potassium phosphate buffer (0.1 M of KH2PO4, 0.1 M of K2HPO4, 530 nM of O-diansidine and 0.0005% of H2O2)] and read at 490 nm.
Bronchoalveolar lavage protein assay
The acquisition of bronchoalveolar lavage (BAL) was done as previously described.20,21 This lavage fluid was centrifuged at 1500 g for 10 min at 4℃ for a protein concentration assay.20,21
Plasma cytokine measurement by cytometric bead array
Blood was collected via cardiac puncture into heparinized syringes, centrifuged, and plasma samples were collected and stored at –70℃ for cytokine analysis. Mice TNF-α, MCP-1, IL-10 and IL-6 levels in plasma were determined by using the cytometric bead array technique (BD Cytometric Bead Array Mouse Inflammation Kit; BD Biosciences). Procedures were carried out according to the manufacturer’s instructions.
Lung tissue cytokine measurement by ELISA
Lung tissue samples were homogenized in the lysis buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 10 mM NaPhosphate, 10 mM NaF, 1 mM Na-orthovanadate, 0.5% TritonX100 and protease inhibitor cocktail) for 30 s, kept on ice for 30 min, centrifuged and lysate collected. Protein content was determined by a standard protein assay using BIO-RAD (Hercules, CA, USA) reagents. Mouse KC, MIP2, IL-6, MCP-1, IL-10 and TNF-α levels in lung tissue were determined with ELISA (BD Bioscience) as described previously.19–21
Immunohistochemical staining for assessment of neutrophil influx and tissue architecture
For histological assessment, the trachea was cannulated, and lungs were gently inflated with 10% formalin through the cannula by a continuous release pump under pressure and volume-controlled conditions (12 ml/h; 5 min) and then paraffin embedded. Samples were then stained with hematoxylin and eosin, and examined by light microscopy for lung morphology. Staining for leukocyte-specific esterase, naphthol AS-D chloroacetate esterase (Sigma Diagnostics, St Louis, MO, USA) was performed on tissue sections fixed in citrate-acetone-formaldehyde.20,21 Slides were incubated in a solution of sodium nitrate, fast red violet BL base solution, TRIZMAL 6.3 buffer and naphthol AS-D chloroacetate solution in deionized water for 15 min at 37℃. After rinsing, slides were counter-stained with Gills hematoxylin solution and cover-slipped. Stained lung sections were examined microscopically for morphology and positively-stained cells. To establish the percentage (%) of cells (per field) that were neutrophils (esterase+ and a unique pattern of punctate staining) present in the sample, tissue sections were randomly screened (7–8 fields/slide) at 400 × (25 µm2/field).20,21
Statistical analysis
Data were expressed as mean ± SEM and analyzed using GraphPad Prism 5 statistical analysis and graphing software. Unpaired Student’s t-test was used to determine differences between the two groups according to Microsoft Excel. Multiple group comparisons were performed using one-way AVONA with the post hoc test of Tukey's. P < 0.05 was considered significant.
Results
TAT-SNAP-23 inhibits TNF-α-induced priming of mouse blood neutrophil respiratory burst in vitro
It has been shown in vitro that blocking human neutrophil granule exocytosis by TAT-SNAP-23 inhibited TNF-α-induced priming of the respiratory burst. 33 In the current study, we wanted to test the effect of TAT-SNAP-23 in a mouse model of in vivo ‘priming’ by Hem followed by septic challenge. First, we performed a dose response to determine an effective concentration of TAT-SNAP-23 using neutrophils isolated from naive mice. Mouse neutrophils were pretreated with TAT-SNAP-23 at 0.4 µg/ml and 0.6 µg/ml for 10 min, then respiratory burst capacity following TNF-α priming was measured using PMA as the stimulus. The results showed that 0.6 µg TAT-SNAP-23/3 × 105 cells/ml was effective to inhibit respiratory burst induced by TNF-α priming (Supplementary Figure 1).
In vivo administration TAT-SNAP-23 in mice alters ex vivo respiratory burst capacity of blood neutrophils from mice subjected to experimental septic challenge or hypotensive shock
CLP model
After establishing that TAT-SNAP-23 was able to inhibit the TNF-induced respiratory burst in vitro using mouse neutrophils—as it has been shown that the cells are not only affected by sepsis, but are proposed to serve as mediators (via dysfunctional regulation of their respiratory burst function) of by-stander organ injury seen in sepsis—we hypothesized that TAT-SNAP-23 should have a role in regulating neutrophil respiratory burst induced by septic challenge in vivo. To address this hypothesis, a preliminary study was conducted in an attempt to establish the optimum time point for assessing neutrophil burst activity after CLP. We found that we could detect little priming for respiratory burst activity at either 12 or 24 h after CLP; however, at 4 h after CLP a relatively good respiratory burst was detected (data not shown). Subsequently, the CLP mice were divided into two groups with or without treatment of 6 or 12 µg/mouse of TAT-SNAP-23 (10 or 20 times the in vitro dose). Four h after surgery, blood neutrophils were isolated and respiratory burst was tested similarly to the in vitro studies described earlier. Figure 1 shows that PMA stimulated respiratory burst capacity was significantly inhibited by 12 µg/mouse dose of TAT-SNAP-23, but not by 6 µg/mouse.
In vivo dose response of TAT-SNAP-23 in CLP model. CLP mice were given TAT-SNAP-23 (6 or 12 µg) in 200 µl HBSS/mouse, or vehicle (200 µl HBSS/mouse) by i.v. tail injection right after surgery. Blood was collected 4 h after surgery. Neutrophils were isolated and re-suspended in 100 -µl sterile HBSS, then added to 100 µl of 0.398% cytochrome C and 20 nM of PMA (as activating agonists) for burst test. The blank control was set as no cells; the negative control was set as without 20 nM of PMA. Sham CLP mice were given 6 µg TAT-SNAP-23 in 200 µl HBSS/mouse and neutrophils were also isolated for respiratory burst test as baseline. Sham mice had small/no neutrophil respiratory burst test. The dose of 12 µg/mouse TAT-SNAP-23, not 6 µg/mouse, significantly inhibits the neutrophil respiratory burst capacity in CLP mice (n = 6–10/group, *P < 0.05 12 µg/mouse versus sham CLP, HBSS/vehicle and 6 µg/mouse CLP group, determined by one-way AVONA for multiple group comparisons). A typical result of three groups is provided.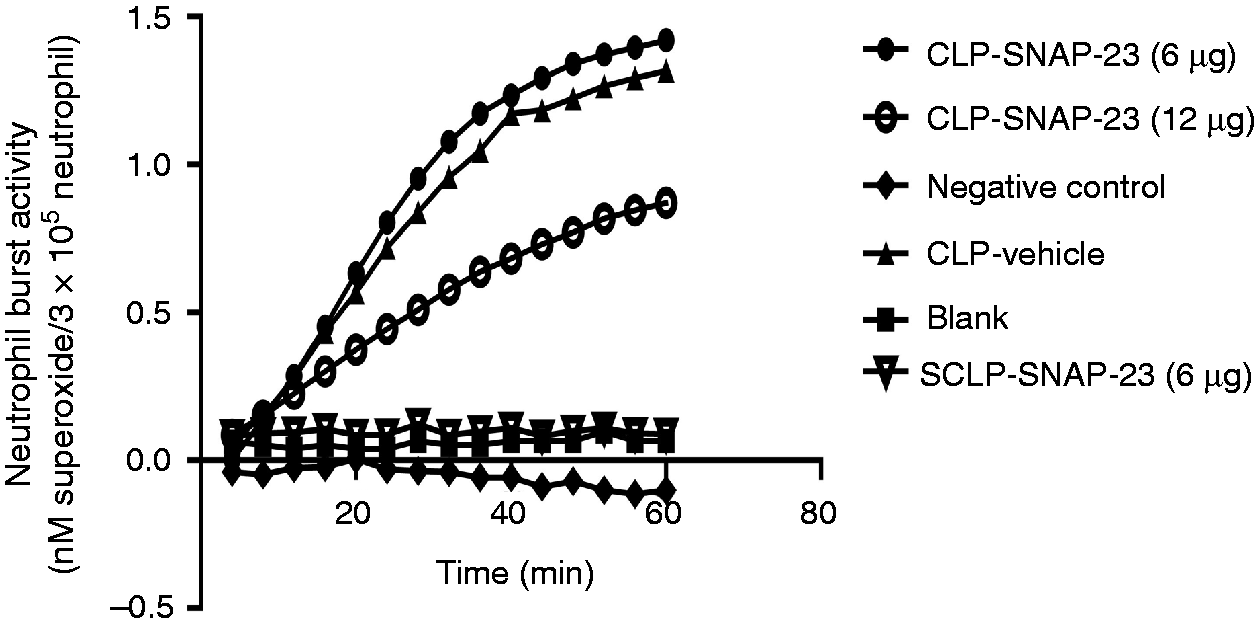
Hem alone model
In previously published experiments we have shown that non-lethal Hem serves to transiently prime for enhanced respiratory burst capacity of neutrophils, which appears to contribute to the induction of lung injury in response to a subsequent septic challenge.
19
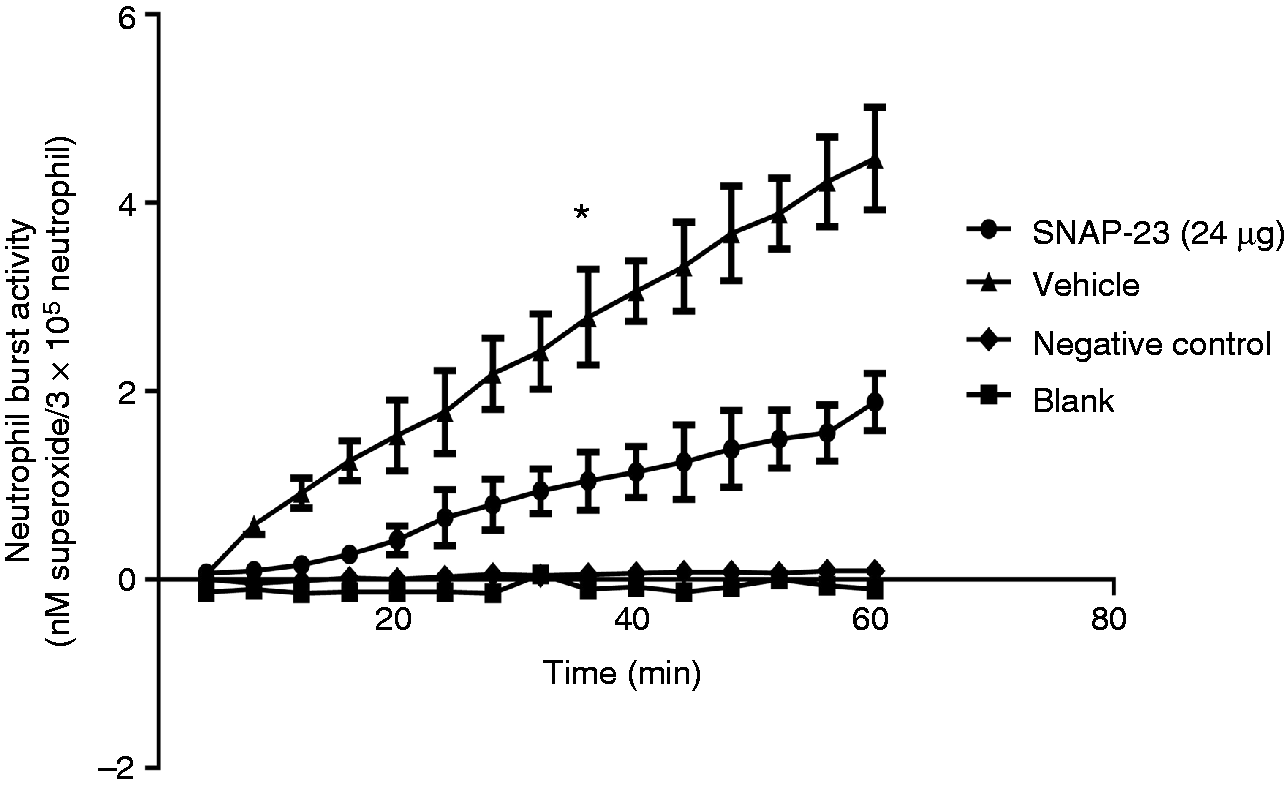
To determine if i.v. administration of TAT-SNAP-23 inhibited Hem-induced priming of the neutrophil respiratory burst, we first set out to establish the kinetics of the change in ex vivo blood neutrophil respiratory burst capacity over time 4, 12 and 24 h after Hem/resuscitation. Figure 2 shows that there is no priming of PMA-stimulated superoxide release 4 h after Hem; however, when cells were taken for assessment at 12 h, a priming response is observed after PMA stimulation, which is reduced if cells are obtained at 24 h. These data indicate that the 12-h time point is the optimum time for assessing the in vivo capacity of TAT-SNAP-23 administration to affect neutrophil priming ex vivo. That said, when we initially examined the in vivo capacity of 12 µg/mouse (the effective dose that altered septic mouse blood neutrophil rise in ex vivo respiratory burst capacity) of TAT-SNAP-23 we found that this dose in Hem mice (data not shown) only slightly decreased the respiratory burst capacity, while 24 µg/mouse (Figure 3) significantly (P < 0.05) inhibited the respiratory burst comparing them with vehicle treatment group. Based on these results, 24 µg/mouse TAT-SNAP-23 was selected for use in the following studies using our Hem-priming followed by CLP model for the development of iALI in mice.
Optimal time points for neutrophil respiratory burst testing after Hem alone. Blood was collected at three different timepoints (4, 12 and 24 h) after Hem. Neutrophils were isolated and re-suspended in 100 µl sterile HBSS, then added to 100 µl of 0.398% cytochrome C and 20 nM of PMA (as activating agonists) for burst test. The blank control was set as no cells; the negative control was set as without 20 nM of PMA. Twelve h after Hem is the optimum time to give TAT-SNAP-23. A typical result of three independent experiments is provided. TAT-SNAP-23 inhibits neutrophil respiratory burst in Hem model. Hem mice were given 24 µg TAT-SNAP-23 in 200 µl HBSS/mouse or vehicle (200 µl HBSS/mouse) by i.v. infusion during resuscitation period. Blood was collected 12 h after surgery. Neutrophils were isolated and re-suspended in 100 µl sterial HBSS, then added to 100 µl of 0.398% cytochrome C and 20 nM of PMA (as activating agonists) for burst test. The blank control was set as no cells; the negative control was set as without 20 nM of PMA. Twenty-four µg/mouse TAT-SNAP-23 significantly inhibits the neutrophil respiratory burst 12 h after Hem compared with vehicle-treated control group (n = 6–8/group, *P < 0.05 TAT-SNAP-23 versus vehicle treatment group, determined by unpaired Student’s t-test). All data are expressed as mean ± SEM.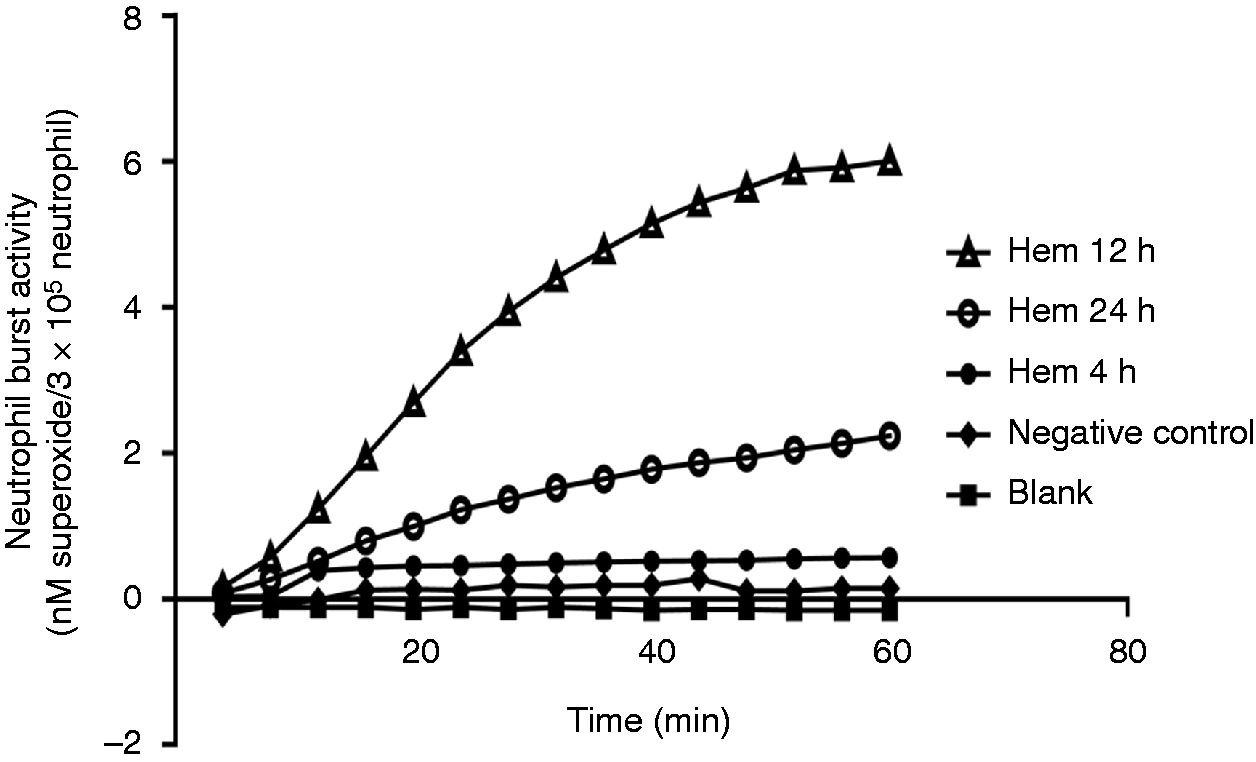
Neutrophil influx to the lungs is significantly blocked by in vivo treatment with TAT-SNAP-23
In both the CLP and the iALI models, TAT-SNAP-23 significantly inhibited PMA-induced respiratory burst capacity. We next wanted to evaluate if inhibition of respiratory burst capacity affected neutrophil infiltration into the lungs. Our data indicate that neutrophil influx, as assessed by MPO activity, was significantly reduced in lung tissue of CLP mice that received 12 µg/mouse TAT-SNAP-23 after 4 h of treatment (Figure 4A). Figure 4A shows that TAT-SNAP-23 treatment significantly decreased tissue MPO activity by 48% when compared with control mice in the iALI model (Hem/CLP). Supporting the MPO data, lung tissue histology from both CLP and iALI mice that received TAT-SNAP-23 showed a significant reduction in the percentage of neutrophil-specific esterase+ cells compared with their respective vehicle treatment control group (Figure 4B).
Neutrophil influx significantly blocked by TAT-SNAP-23. Lung tissue MPO activity (A), a measure of neutrophil influx to lung, was reduced in lungs from both CLP and iALI model compared with respective control groups (n = 6–8/group, *P < 0.05 TAT-SNAP-23 versus vehicle treatment CLP group; TAT-SNAP-23 versus vehicle treatment iALI group, determined by unpaired Student’s t-test). (B) This is consistent with the percentage of esterase+ (neutrophil-specific) cells compared with lung tissue from vehicle treatment control group (n = 4–8/group, *P < 0.05 versus equivalent vehicle treatment group, @P < 0.05 TAT-SNAP-23 treatment iALI group versus TAT-SNAP-23 treatment CLP group, determined by unpaired Student’s t-test). All data are expressed as mean ± SEM. Dashed line represents sham Hem/sham CLP (SHem/SCLP) baseline level.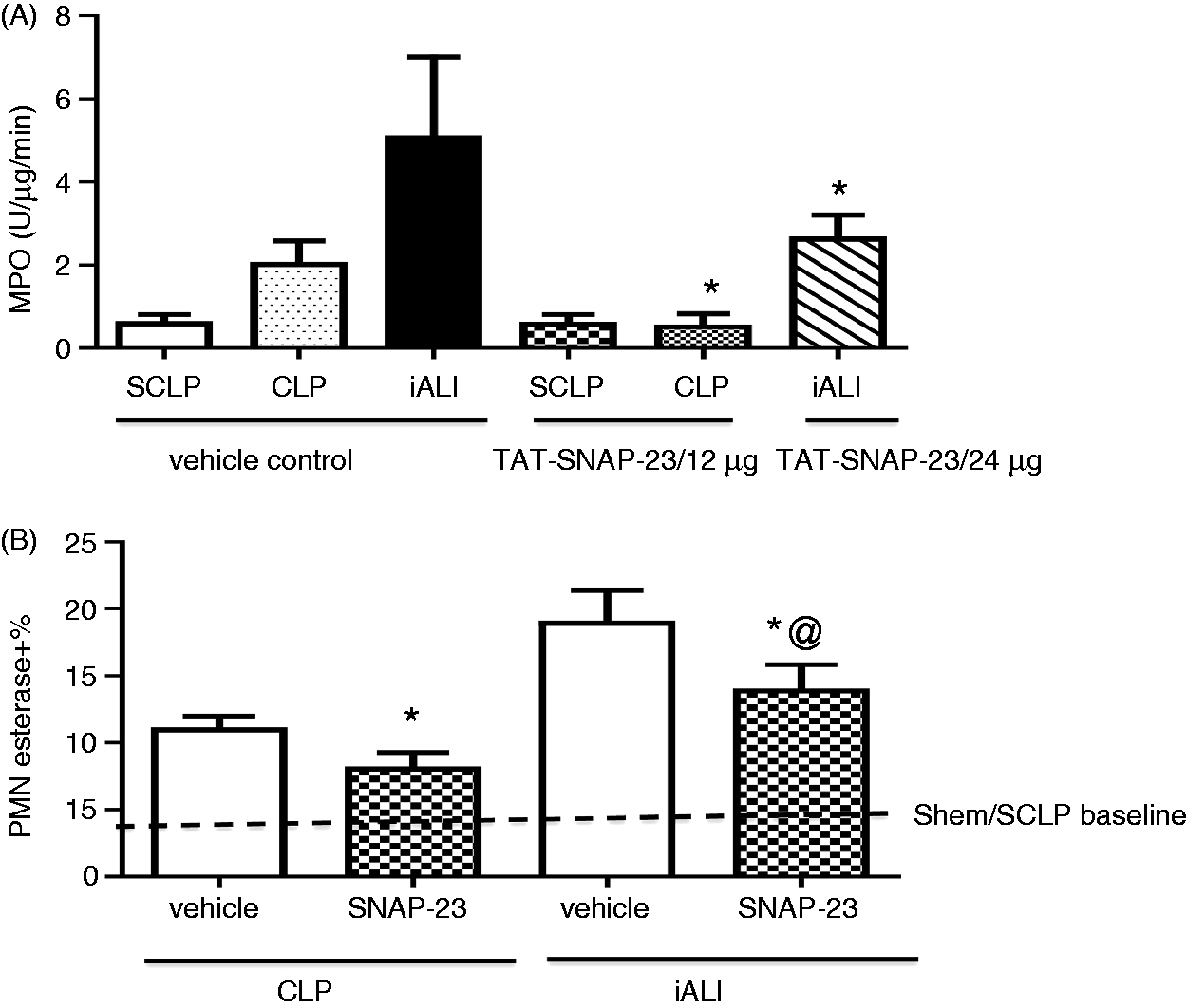
In vivo TAT-SNAP-23 administration suppresses ex vivo neutrophil migratory capacity
To further investigate the potential mechanisms by which inhibition of neutrophil influx was induced by TAT-SNAP-23 as seen in the data given above (Figure 4), blood neutrophils were isolated at 4 h post-CLP, or 24 h post-Hem alone, and their corresponding control groups were evaluated ex vivo for their ability to migrate toward classical chemotactic agent fMLP (20 µM) using the modified Boyden chamber in vitro assay. As shown in Figure 5, neutrophil migration from control CLP mice was 88 ± 12 cells/field, and this was significantly reduced in the TAT-SNAP-23 treated animals (48 ± 8 cells/field). Similarly, neutrophil migration from control Hem mice was 48 ± 8 cells/field, and TAT-SNAP-23 treatment significantly reduced neutrophil migration (26 ± 6 cells/field). The significant reduction in neutrophil migration in both mouse models treated with TAT-SNAP-23 is consistent with the MPO data. These data indicate that TAT-SNAP-23 treatment blunted the in vitro chemotactic capacity of neutrophils isolated from CLP and Hem mice.
Effects of TAT-SNAP-23 on neutrophil chemotaxis ability in vitro. Isolated neutrophils (1 × 104 cells) from CLP and Hem mice with or without treatment with TAT-SNAP-23 were pipetted into the top wells of a modified Boyden chamber, while the DMEM medium with fMLP (20 µM) was placed in the lower chambers. Migration was carried out for 1 h. After incubation, the number of migrated cells was counted at 100 × magnification on at least five random fields. Each datum represents the mean ± SEM from four experiments in each group (n = 4/each group, * P < 0.05 TAT-SNAP-23 treatment groups versus equivalent vehicle treatment control groups, determined by unpaired Student’s t-test). All data are expressed as mean ± SEM.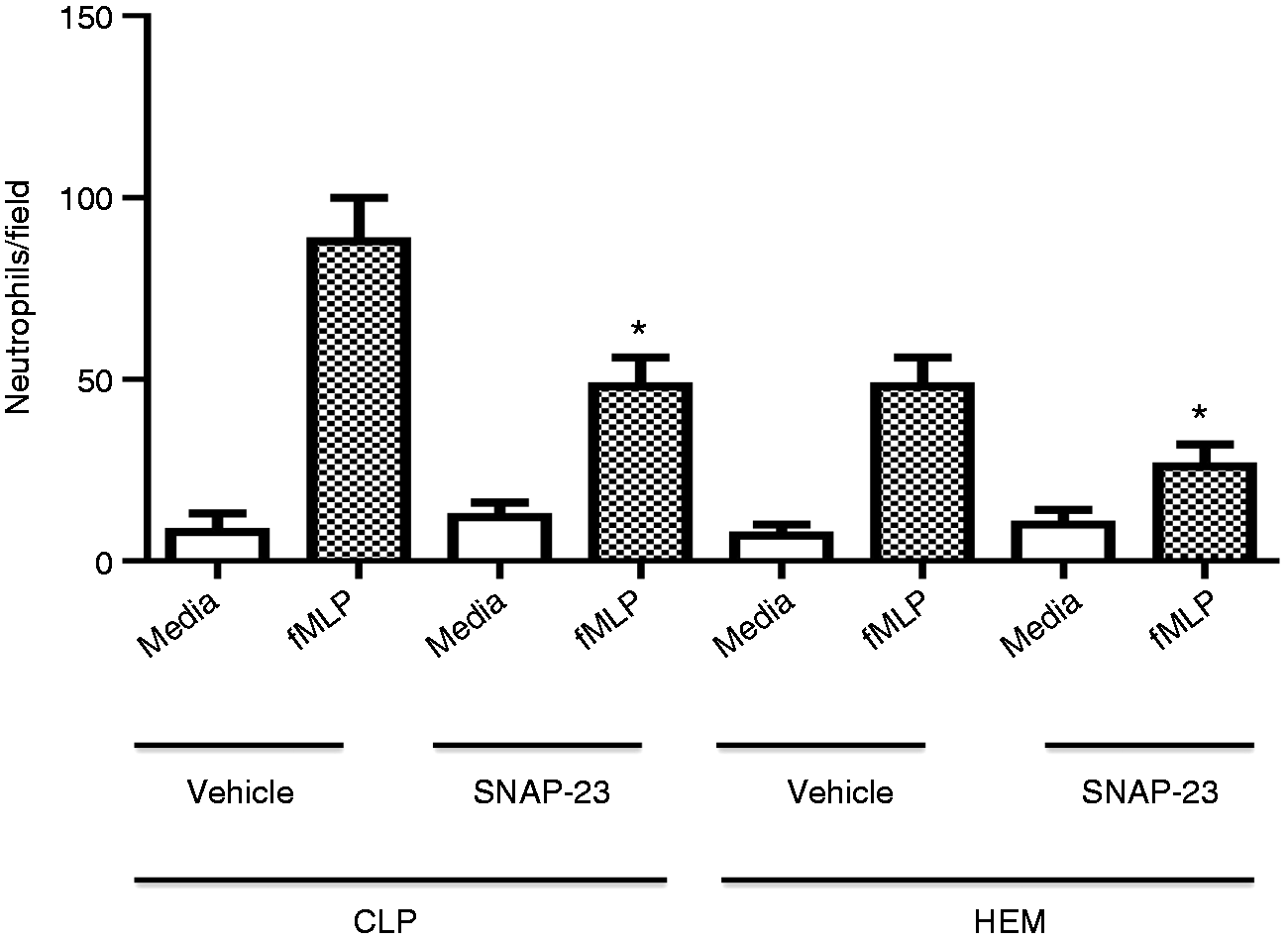
In vivo TAT-SNAP-23 treatment preserves lung architecture in iALI mice
To the extent that the administration of TAT-SNAP-23 decreased the neutrophil infiltration into the lungs of iALI mice, we further wanted to know whether TAT-SNAP-23 would reduce the degree of lung injury. Figure 6 shows representative lung histology from iALI mice with or without treatment of TAT-SNAP-23. Disruption of lung tissue architecture, cellularity and septal thickening were reduced in lung tissue sections from TAT-SNAP-23-treatment mice subjected to Hem/CLP. Histologic changes were less evident in the CLP mice sections compared with their respective controls (data not shown); this is consistent with our former findings and those of Iskander et al.,
35
that is, that CLP alone is not a strong inducer of ALI.
19
TAT-SNAP-23 treatment preserves lung architecture in iALI mice. Hematoxylin and eosin stain of representative sections (100 × magnification) of the lungs of sham Hem/sham CLP (SHem/SCLP) group (A), vehicle control (B) and TAT-SNAP-23 (C) treatment mice 24 h after iALI. Histological findings confirmed that TAT-SNAP-23 significantly reduced lung tissue septal thickening and cellular infiltrate of mice subjecting to iALI (C) compared with vehicle treatment control group (B) and SHem/SCLP group (A).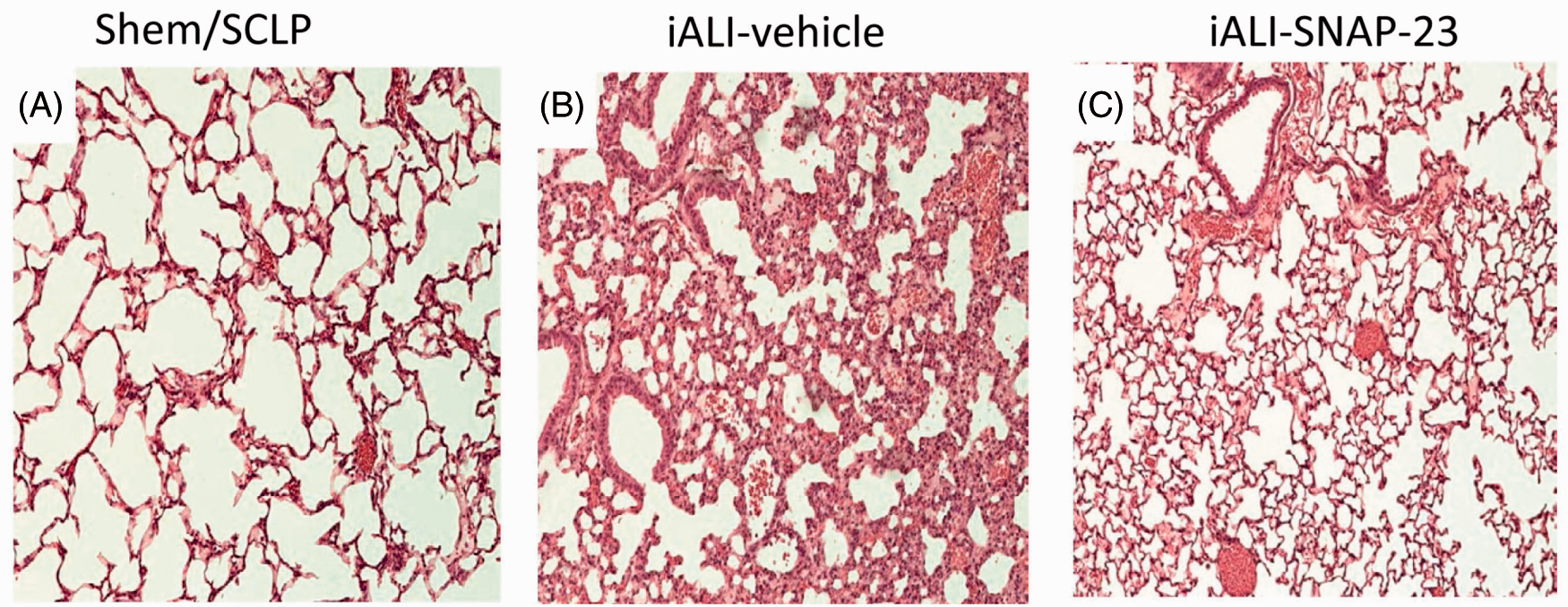
To quantify the amelioration of increased vascular permeability observed in the iALI mice treated with TAT-SNAP-23, protein concentration in BAL fluids were assessed in iALI groups. The protein levels in BAL from TAT-SNAP-23-treated animals were significantly reduced in comparison with the control group (Figure 7). This was consistent with our histological findings of a reduction in the disruption of lung tissue architecture and congestion seen in the TAT-SNAP-23 treatment group (Figure 6B, C).
Protein concentration in BAL fluid from TAT-SNAP-23-treated iALI mice decreased significantly compared with control group (n = 6–8/group, *P < 0.05 TAT-SNAP-23 versus vehicle treatment control iALI group, determined by unpaired Student’s t-test). All data are expressed as mean ± SEM.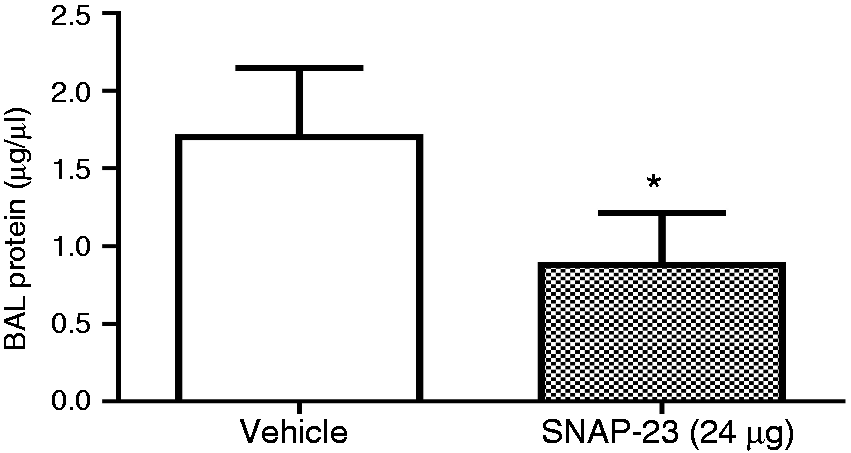
In vivo TAT-SNAP-23 administration has no effect on blood or lung tissue neutrophil chemotactic protein and inflammatory cytokine levels in CLP or iALI mice
Administration of TAT-SNAP-23 had no effect on neutrophil chemokines production in lung in both CLP and iALI model.
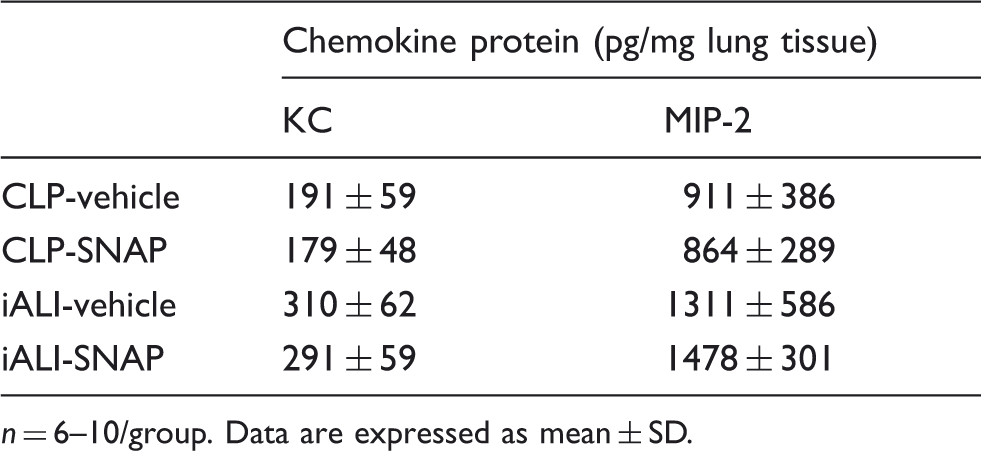
n = 6–10/group. Data are expressed as mean ± SD.
Administration of TAT-SNAP-23 had no effect on inflammatory cytokines release in both the CLP and iALI models.
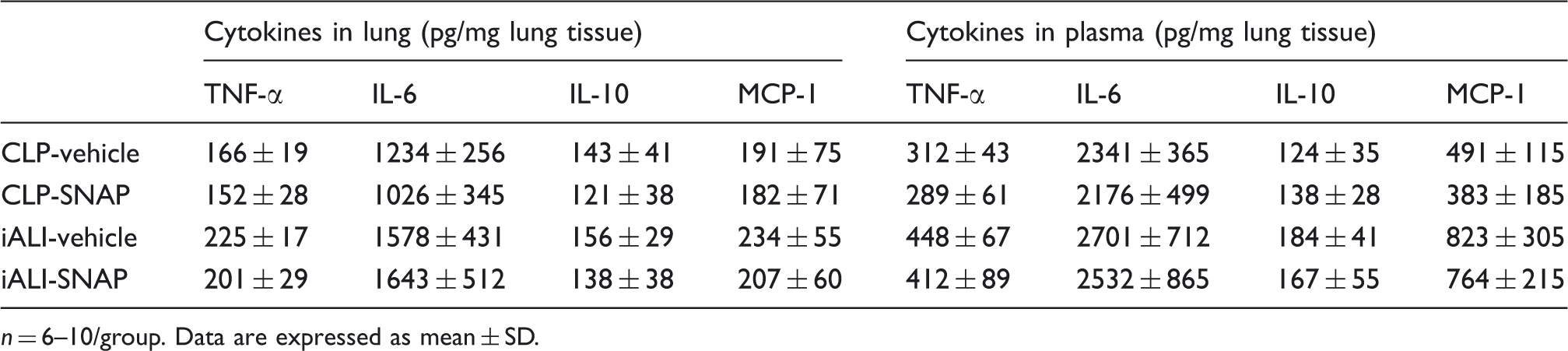
n = 6–10/group. Data are expressed as mean ± SD.
Discussion
Neutrophils contribute to the primary cellular defense against bacterial and fungal infections. Neutrophils ingest microbes by a process known as phagocytosis, and the ingested microorganisms are destroyed by the combination of ROS and cytotoxic granule components. 36 Martins et al. 37 have reported that generation of NO and ROS by neutrophils was increased in septic patients, and their persistence was associated with poor outcome. 38 Unfortunately, no effective drug is currently available that controls and terminates the neutrophil’s burst activity and prevents neutrophil-dependent inflammation without compromising other aspects of the innate immune response.
In this respect, SNARE proteins could represent just such a potential selective inhibitor of neutrophil function, as these proteins play a central role in intracellular membrane trafficking by mediating fusion of membranes from different cellular compartments of these cells.28–30 This, in turn, affects processes related to neutrophil function, such as granule exocytosis, migration and/or respiratory capacity. Interestingly, it has been reported that inhibition of SNARE protein function with TAT-SNAP-23 was able to selectively block neutrophil degranulation and reduced TNF-mediated respiratory burst priming in human neutrophils. 33 However, until now, no studies have focused on these proteins or the effect of TAT-SNAP-23 inhibition of SNARE proteins on the sepsis-induced neutrophil respiratory burst response. To address this, we hypothesized that TAT-SNAP-23 would decrease sepsis-induced neutrophil respiratory burst in an experimental model of this condition. In this study, we show for the first time that in vivo septic challenge priming of neutrophil respiratory burst capacity is significantly inhibited by the intravenous administration of TAT-SNAP-23 compared to vehicle-treated controls.
Based on these data, we sought to further investigate the role of TAT-SNAP-23 in Hem-induced priming for neutrophil respiratory burst capacity (as this is thought to set the stage for the development of indirect acute lung injury similar to that seen in critically injured trauma patients with ARDS) when the animal is subjected to a secondary inflammatory/infectious challenge like peritoneal sepsis.19–21,23,24 According to our previous studies, peripheral blood neutrophils from C3H/HeN mice isolated 24 h after Hem (Hem alone), but not sham Hem, exhibited an increase in ex vivo respiratory burst capacity, consistent with in vivo ‘priming’ for iALI. 19 As we used a different strain of mice (C57BL/6) to test the respiratory burst in this study, it was important to reassess the kinetics of optimal neutrophil priming following shock. The time course results show that, importantly, while there was still an increase in ex vivo respiratory burst capacity of blood neutrophils isolated from these C57BL/6 mice at 24 h post-Hem, the peak increase in capacity was seen at 12 h post-Hem. Consistent with the result from CLP model, we also found that in vivo TAT-SNAP-23 treatment of mice significantly inhibited their ex vivo neutrophil respiratory burst capacity when assessed at 12 h post-Hem.
Having documented that TAT-SNAP-23 significantly decreased the ex vivo blood neutrophil respiratory burst both in CLP and Hem alone mice, we sought to further investigate the role of TAT-SNAP-23 in our dual-insult (Hem priming subsequent to septic challenge) model of iALI. We found that lung tissue sections from the TAT-SNAP-23-treated iALI group showed significantly less disruption of normal lung tissue architecture, including reduced tissue congestion. In addition, BAL fluid protein concentration (an index of lung injury/permeability) from the TAT-SNAP-23-treated group was reduced compared with the vehicle-treated control group. This observation is supported, in part, by the recent findings of Uriarte et al., 34 using an immune complex mediated deposition direct lung injury model in rats, in which they showed that TAT-SNAP-23 treatment resulted in a reduction in lung edema, vascular Hem, vascular permeability, total BAL fluid protein levels and a decline in lung tissue MPO. We have further broadened the potential treatment indications of TAT-SNAP-23 by demonstrating protection against lung injury of indirect causes (hemorrhagic shock/polymicrobial sepsis) in our mouse model, which is thought to better reflect the clinical scenario of ARDS encountered in the severely injured patients and pathophysiologically distinct from a direct insult to the lung.9–12
In the current study, we found that MPO (an index of neutrophil influx) levels in lung tissue were decreased in TAT-SNAP-23-treated mice compared with the vehicle-treated controls. Consistent with the reduced tissue MPO levels, we found that TAT-SNAP-23 treatment also suppressed the number of neutrophil-specific esterase staining cells seen in lung tissue sections from both CLP and iALI mice. Morrell et al. 39 have shown that intravenous administration of a TAT fusion protein containing an inhibitory peptide derived from NSF inhibited vascular endothelial cell exocytosis and blocked neutrophil infiltration into the peritoneum in a mouse model of abdominal sepsis. These findings support our results showing that administration of TAT-SNAP-23 reduced the neutrophil influx into lungs both in septic insult and iALI. Our finding of reduced neutrophil recruitment differs from the work of Uriarte et al., 34 who indicated that neutrophil infiltration is not affected by TAT-SNAP-23 treatment following immune complex deposition in a rat ALI model, which is thought to be dependent on complement activation, cytokine production, and/or chemokine-induced neutrophil infiltration. One possible explanation for the difference could be that we used a two-hit mouse model where the indirect-pulmonary insults of Hem-induced neutrophil priming is followed by peritoneal (another indirect-lung insult19–21,23,24) polymicrobial septic challenge, which may be pathophysiologically distinct from those changes seen in response to immune complex deposition induced (direct pulmonary insult) ALI in rats.
Neutrophils are the earliest immune cells to be recruited to the site of injury/inflammation, and their activation and recruitment are thought to play key roles in the progression of iALI and sepsis-associated organ injury/dysfunction.40,41 Here, we sought to further investigate the potential mechanism by which targeting neutrophil SNARE proteins with TAT-SNAP-23 affect neutrophil influx. A number of studies in mice have provided evidence supporting the concept that the most relevant chemokines for neutrophil recruitment into lungs are KC and MIP-2.24,42,43 Further, our previous Ab inhibition studies have shown that anti-MIP-2α, but not anti-KC, treatment reduced neutrophil recruitment into lungs in our iALI model. 20 It has also been reported by other laboratories that SNAP-23 is required for the release of all chemokines by mature human mast cells and the regulation of macrophage adhesion, spreading and migration on fibronectin.44–47 In light of this, we sought to assess whether the affect on neutrophil influx following in vivo treatment with TAT-SNAP-23 could be explained by its direct/indirect effect on lung tissue or circulating levels of these two chemokines. However, our data showed that in vivo treatment with TAT-SNAP-23 did not reduce either lung tissue KC or MIP-2 levels in either the CLP or iALI models.
Interestingly, we found, irrespective of the inability to affect these chemokine levels, that TAT-SNAP-23 treatment markedly blunted the ex vivo migratory capacity of neutrophils from both the septic-challenged and Hem-alone mice. These findings may provide a partial explanation for the capacity of TAT-SNAP-23 treatment to reduce neutrophil influx to lungs of these mice.
Four distinct phases of neutrophil migration have been identified: mobilization, margination and rolling, adherence, and transmigration through the vessel wall in response to chemotactic gradients, all of which are affected by inflammatory activation.40,48 And while multiple mechanisms have been shown to contribute to sepsis and ALI/ARDS-induced impairment of neutrophil migration, these processes are complex and our understanding remains incomplete.40,48 It has been reported that injurious oxidants increase endothelial and/or epithelial cell permeability via disruption of tight junctions and redistribution of junction proteins during neutrophil transmigration. 40 This is thought to be a key factor in the pathogenesis of sepsis and ALI;40,49 thus, neutrophils are thought to be associated with high levels of plasma (vascular permeability) and lung oxidants (neutrophil respiratory burst).50,51 As previous studies have confirmed TAT-SNAP-23’s effects on the inhibition of exocytosis, reduction of the NADPH oxidase activity and production of ROS,33,52 whether TAT-SNAP-23 has some effect on the neutrophil migration into the lung through a process dependent on oxidant-induced junction disruption remains to be determined in future experiments.
In this study, we first showed that TAT-SNAP-23 has an effect on reducing neutrophil recruitment and/or migration in turn suppressing injury to the lung in a model of indirect/extra-pulmonary insult that is believed to more closely mirror the clinical scenario associated with critically ill patients. However, TAT-SNAP-23 did not affect the cytokine response to sepsis and iALI. This observation is supported, in part, by the recent findings in a rat ALI model, in which TAT-SNAP-23 had no marked effect on the cytokine production and release by resident macrophages, lung parenchymal cells or infiltrating neutrophils. 34
In summary, our data demonstrate that administration of TAT-SNAP-23 resulted in a significant reduction of neutrophil respiratory burst capacity in sepsis and Hem, with concomitant amelioration of tissue damage in Hem-primed iALI, and decreased neutrophil recruitment into lung in both CLP and indirect ALI mice. Activated neutrophils mount a vicious respiratory burst that contributes to the initiation of iALI by increasing both the inflammatory response (neutrophils recruitment) and lung (microvascular endothelial and epithelial cells) permeability in our animal model. Our results shed more light on the potential mechanisms of neutrophil-dependent complications such as in sepsis and iALI/ARDS. However, the significance of these animal experimental studies concerning the role of neutrophil respiratory burst activity in inflammatory and infectious diseases has been hampered by a large variability of clinical results. Some studies have shown increased ROS release,37,53 whereas others have exhibited reduced ROS release.54,55 However, more recently, a study has reported a threefold increase in release of ROS by circulating activated neutrophils using a human model of controlled systematic inflammation. 56 Furthermore, more clinical data about the neutrophil burst activity in these diseases are needed before extrapolating our data to the development of future concepts for therapeutic remedies for sepsis and/or iALI.
Footnotes
Funding
We acknowledge funding from NIH grants R01GM107149 (to A.A.), K99/R00 HL087924 (to S.M.U.) and BX001838 from the Department of Veterans Affairs (to K.R.M.).
Acknowledgements
We thank Mr. Paul Monfils at the Core Laboratories Facilities at Rhode Island Hospital for the assistance with the histological preparation/staining done in this study.
Conflict of interest
The authors do not have any potential conflicts of interest to declare.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.