
Abstract
Proper development and activation of cells of the myeloid lineage are critical for supporting innate immunity. This myelopoiesis is orchestrated by interdependent interactions between cytokine receptors, transcription factors and, as recently described, microRNAs (miRNAs). miRNAs contribute to normal and dysregulated myelopoiesis. Alterations in myelopoiesis underlie myeloid-derived suppressor cell (MDSC) expansion, a poorly understood heterogeneous population of immature and suppressive myeloid cells that expand in nearly all diseases where inflammation exists. MDSCs associated with inflammation often have immunosuppressive properties, but molecular mechanisms responsible for MDSC expansion are unclear. Emerging data implicate miRNAs in MDSC expansion. This review focuses on miRNAs that contribute to myeloid lineage differentiation and maturation under physiological conditions, and introduces the concept that altered miRNA expression my underlie expansion and accumulation of MDSCs. We divide our miRNAs into those with potential to promote MDSC expansion and two with known direct links to MDSC expansion, miR-223 and miR-494.
Introduction
Myelopoiesis generates innate immune cells, such as monocytes, granulocytes and dendritic cells.1–3 Under physiological conditions, monocytes and granulocytes derive from multipotent hematopoietic progenitors through sequential events of lineage restriction, differentiation and maturation.4,5 This process is orchestrated by interactions among cytokine receptors, transcription factors and microRNAs (miRNAs). 6 These interactive regulatory processes may, under certain conditions, alter myeloid cell differentiation and maturation, leading to myeloproliferative disorders. For example, in acute myeloid leukemia (AML) the myeloid maturation program is arrested, resulting in accumulation of immature myeloid progenitors.7–9 A growing concept is that miRNAs support development, maintenance and regulation of normal and abnormal myelopoiesis.5,8,10–12 A poorly understood outcome of distorted myelopoiesis is generation of myeloid-derived suppressor cells (MDSCs).
MDSCs
MDSCs are a heterogeneous population of immature myeloid cells (see Figure 1).13–15 Although immature myeloid cells with suppressive function were described ∼20 yrs ago as ‘natural suppressor cells’,16–18 they have been called MDSCs only recently.
19
They are generated in nearly all acute and chronic inflammatory conditions,13,14,19 but their repressor properties are best characterized in murine models of cancer, where they disrupt T-cell-dependent anti-tumor properties.13,14 Recently, there have been reports that MDSCs expand during murine sepsis.20,21
A schematic of myeloid lineage development and differentiation depicting how changes in the expression of some miRNAs can disrupt myeloid progenitor differentiation and generate MDSCs. MDSCs may arise before or after the GMP stage, as they exist at different myeloid progenitor and precursor stages. Not all miRNAs suspected to regulate MDSC biology are shown; only those that have been investigated directly in MDSCs are shown. The lymphoid and erythroid differentiation paths are not discussed in this review and thus are not shown. HSC: hematopoietic stem cell; MPP: multipotent progenitor; LMPP: lymphoid-primed multipotent progenitor; CMP: common myeloid progenitor; GMP: granulocyte-macrophage progenitor.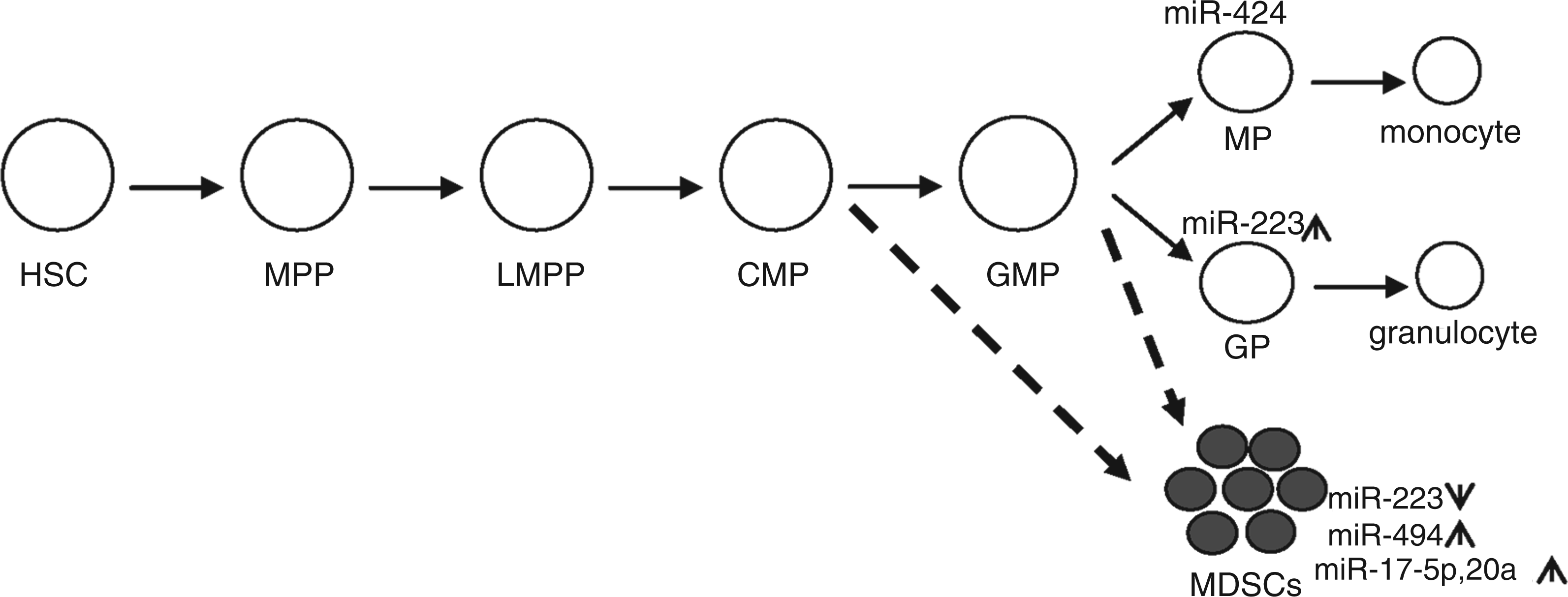
Immature myeloid cells are precursors of monocytes, granulocytes and dendritic cells. Under certain pathophysiological conditions, such as a growing tumor or an infection, these immature myeloid cells are arrested at different stages of maturation.13,14 It is suggested that the tumor or inflammation microenvironments modify terminal differentiation, resulting in accumulation of MDSCs. Thus, MDSCs are not necessarily a distinct subset but rather a heterogeneous population of activated immature myeloid cells.13,14,19 As such, it is not surprising that some MDSCs also express markers that are retained on more mature monocytes and neutrophils, such as Ly6C and Ly6G respectively. Murine MDSCs are classified by the expression of surface markers CD11b and Gr1 (myeloid lineage differentiation antigen), the latter being comprised of two antigens: Ly6C and Ly6G.13,22 Based on the relative expression of these two Ly6 antigens, MDSCs are further divided into monocytic (CD11b+ Ly6G+/− Ly6Chi) and granulocytic (CD11b+ Ly6Ghi Ly6C−) subsets.22–24 Both subsets can express pro- and anti-inflammatory mediators.22,25,26 For example, Delano et al. 26 showed that MDSCs expanding during murine sepsis produce pro-inflammatory cytokines, such as TNF-α and IL-1β, and anti-inflammatory/immunosuppressive cytokines, such as IL-10, when stimulated ex vivo with bacterial endotoxin/LPS. Recent studies showed that repressor MDSC of tumor-bearing mice have increased expression of NO synthase and arginase, which suppress T-cell proliferation resulting in enhanced tumor progression.24,27 However, MDSCs in other pathological conditions have heterogeneous function, which protect 21 or suppress the immune response.19,28,29 We reported that MDSCs suppress the immune response and contribute to the late immunosuppression observed in murine sepsis with increased IL-10 and arginase expression, 20 whereas MDSCs of early murine sepsis produce pro-inflammatory cytokines TNF-α and IL-6. These findings underscore the phenotypic and functional plasticity of this heterogeneous cell population. Recently, Gabrilovich et al. 22 proposed a model of two overlapping signals that regulate the distinct features of MDSCs. According to this model, a signaling path of STAT3 and proliferation and survival factors, such as c-Myc, Cyclin D and Survivin, promote proliferation, but prevent differentiation and maturation of immature myeloid progenitors. A second signaling path uses STAT3, PI3K and NF-kB transcription factors to induce immunosuppressive mediators, such as arginase, IL-10 and TGF-β. Together, these factors produced at sites of inflammation or in tumor microenvironment may expand MDSCs and/or enhance their immunosuppressive functions.13,14,19
MDSCs exist in different stages of myeloid differentiation. Under physiological stress, such as in response to infection or a growing tumor, there is a need for expansion of the myeloid cell pool in the bone marrow and spleen to compensate for the loss of innate immune cells during inflammatory processes.30,31 This prompted the proposal that immature myeloid progenitors (i.e. MDSCs) may accumulate as components of ‘emergency’ myelopoiesis, 19 or better called ‘inflammatory-induced myelopoiesis’. Factors that prevent their terminal differentiation and maturation increase their accumulation. Although this might explain the increases in numbers of MDSCs, it does not clearly define the factors that prevent MDCS from terminal differentiation or determine whether they are pro-inflammatory or immunosuppressive. One possibility may be that factors promoting generation of MDSCs also prevent their terminal differentiation. 19
Most of the above studies were carried out in murine models of cancer or inflammation. While both mice and humans MDSCs are similar in their phenotypic and functional heterogeneity, human MDSCs do not express the Gr1 marker and thus are most commonly phenotyped as CD11b+ CD14− CD33+ (Siglec-3) cells, which also lack the expression of markers of mature myeloid and lymphoid cells, and the MHC class I molecule human leukocyte antigen (HLA)-DR.13,32 Some investigators identify human immunosuppressive MDSCs as CD11b+, CD33+, CD14−, CD15− or CD11b+, CD33+ and HLA-DR− (reviewed in Cuenca et al. 19 ). Others identify MDSCs in cancer patients as CD33+, CD34+, CD15+ and CD16+ (reviewed in Dilek et al. 33 ). Moreover, a new population of MDSCs phenotyped as CD14+ HLA-DR−/low has been detected in the blood of patients with malignant melanoma 34 and hepatocellular carcinoma. 35 Kusmartsev et al. 32 reported significant increases in circulating MDSCs in patients with metastatic renal cell carcinoma. They further showed that treatment of these cells with all-trans retinoic acid—a metabolite of vitamin A, which has been shown to promote differentiation of immature myeloid cells into macrophages and dendritic cells15,36—abrogated MDSC-mediated immune suppression and improved anti-tumor immunity. 32 In addition, a study using a limited number of patients who died from sepsis showed accumulation of immature myeloid cells in the spleen. 19 Furthermore, some studies observed elevated levels of circulating MDSCs in transplant patients (reviewed in Lees et al. 37 ), suggesting that modulating MDSCs numbers or immunosuppressive functions may be targeted for preventing transplant rejection. 37 These multiple and diverse studies of MDSC underscore the MDSC phenotypic plasticity and the confusion generated by so many varying surface antigens, and emphasize the importance of increasing information about biologic features and identifying what determines plasticity. One candidate group of regulators is miRNAs.
miRNA biology
Structure
miRNAs are small, single-stranded, non-coding RNAs of approximately 22 nucleotides. They are transcribed as long primary transcripts mainly by RNA polymerase II, cleaved into short precursors by the nuclease Drosha and are then exported to the cytoplasm where they are processed into mature miRNAs by the nuclease Dicer; after this, they are loaded into the RNA-induced silencing complex (RISC), which contains Argonaute and other RNA-binding proteins that function as regulatory factors mediating the inhibitory effect of RISC.38–41 miRNAs act mainly as negative regulators of gene expression by directly binding to complementary sequences in the 3′ untranslated regions (3′ UTRs) in their target mRNAs, leading to the repression of protein expression through mRNA degradation and/or translation inhibition.41,42 miRNAs can also exert their repressive function when binding to their cognate sites that are artificially placed in the 5′ UTRs or coding regions. 43
Generation
miRNA biogenesis is regulated at the transcriptional level by cell- and tissue-specific transcription factors, and at the post-transcriptional levels by changes in the activities of proteins that control miRNA processing and maturation, such as the nuclease Dicer.47–49 Interestingly, in the hematopoietic system, these two regulatory processes can be affected by acute or chronic inflammatory signals from infection or cancer. 47 Proteins such as SMAD; arsenate-resistant protein 2 (expressed in proliferating hematopoietic cells); adenosine deaminase (up-regulated during inflammation); and terminal uridylyltransferase 4 are implicated directly in the regulation of miRNA transcript processing and maturation (reviewed in O'Connell et al. 47 ). Changes in the levels and/or activity of these proteins can therefore modulate miRNA expression levels. Several other mechanisms of miRNA expression regulation exist, including epigenetic modifications, and DNA mutation and copy number alterations (reviewed in Deng et al., 50 Wu, 51 and Yu and Cheng 52 ). Epigenetic changes, such as histone methylation and deacetylation and DNA methylation, have been linked to deregulation of miRNA expression, at least in cancer cells.50,52 For example, treatment of several human cancer cell lines with the histone deacetylase inhibitor LAQ824 or the DNA methylation inhibitor 5-aza-2′-deoxycytidine have been shown to alter miRNA expression profiles in these cells.50,52 The expression of miR-223 is silenced following its promoter methylation by the AML1/ETO oncoprotein. 53 In addition, DNA mutations or single nucleotide polymorphism are observed in the primary transcripts of specific miRNAs in chronic lymphoblastic leukemia patients, and have been linked to up- or down-regulation of miRNA expression. While most of these observations were derived from studies in cancer cells, the mechanisms involved are likely to regulate miRNA expression under a variety of cellular stress conditions. Thus, miRNA biogenesis and expression involve multiple layers of regulation, which individually or in combination can lead to profound changes in miRNA levels.
Function
miRNAs inhibit translation of target mRNAs at the initiation, elongation or termination step. Some miRNAs, in the context of the RISC complex, can bind to the 5′-terminal cap structure in the 3′UTR of target mRNA and thus interfere with loading of the translation initiation factor eIF4F and its associated co-factors, which will block translation initiation. 43 miRNAs can also inhibit the target mRNA translation at post-initiation steps by inhibiting translation elongation by preventing loading of ribosomal subunit proteins or by promoting early termination of translation via ribosome ‘drop-off’ (reviewed in Filipowicz et al. 43 ). In addition to translation inhibition, the mechanism of miRNA-mediated mRNA degradation has been described in detail. 43 miRNA, with Ago2 protein, facilitates recruitment of the decay machinery components, such as GW182, CCR4-NOT and DCP1/2, to the target mRNA 3′UTR, which leads to mRNA deadenylation, decapping and, subsequently, degradation. This process takes place in cytoplasmic foci known as p-bodies. Thus, changes in miRNA levels can control basic physiological processes, including cell development, differentiation and homeostasis44–46 by dysregulating expression of protein factors involved in these processes.
miRNAs and MDSC regulation
Summary of miRNAs implicated in myeloid differentiation and with potential to regulate MDSC biology.

HSC: hematopoietic stem cell.
This review focuses on miRNAs that are related to the myeloid lineage differentiation and maturation and thus may play a role in generating MDSCs. I limit my discussion to a few miRNAs known to regulate the monocytic and granulocytic lineage differentiation and maturation, and could contribute to MDSC biology. For a more in depth overview on the role of miRNAs in the development and regulation of the immune system I refer the reader to recent reviews.47,58,61
miRNAs involved in myeloid cell development and may affect MDSC expansion
miR-29a
miR-29a affects myelopoiesis, 8 where its expression is normally down-regulated in committed myeloid progenitor cells in mice and humans, and elevating its levels by ectopic expression in these progenitors promotes cell cycle progression and proliferation, resulting in a myeloproliferative disorder and AML. 8 AML involves a block in terminal differentiation of myeloid cells and accumulation of myeloid progenitors. 9 These results were obtained by transplanting myeloid progenitors overexpressing miR-29a into mice, which showed increased expansion of all myeloid progenitors, but, most frequently, the granulocyte-macrophage progenitor population. 8 Although miR-29a increased progenitor numbers, it did not prevent their differentiation to the granulocytic (Gr1+) or the monocytic (Mac1+) precursors. miR-29a is highly expressed in myeloid progenitors of AML patients, 8 where the mechanism responsible for its effect on myeloid progenitor expansion and the phenotype of AML has been studied. miR-29a targets the acute myeloid leukemia 1 (AML1) protein (the DNA-binding subunit of the transcription factor CBF), which induces receptors for macrophage colony-stimulating factor (M-CSF) and granulocyte colony-stimulating factor (G-CSF) that promote monocyte and granulocyte differentiation. 61 Lower AML1 levels promote myeloid proliferation and survival, thereby maintaining the myeloproliferative state in AML. 9 The precise pathway for miR-29a mediating this effect is unclear, but it may target proteins involved in cell cycle transition and proliferation. For example, the high mobility group protein HBP1 is not expressed in myeloid progenitors lacking miR-29a, which results in cell cycle transition from G1 to G2/S phase in proliferating cells and enhanced survival. 8 Thus, miR-29a may induce myeloid progenitor expansion and affect MDSC formation by targeting unidentified proteins involved in regulating differentiation or cell cycle progression.
miR-146a
miR-146a is widely studied, because it is coupled to NF-kB activation, which participates in various immune processes and in cellular proliferation and activation.55,56 miR-146a is also induced by inflammatory signals that link inflammation to cancer. 55 Elevated levels of miR-146a occur in mouse primary macrophages, neutrophils and dendritic cells after TLR stimulation by LPS. 55 Recently, Baltimore and colleagues55,56,62 implicated miR-146a in myelopoiesis by showing that mice deficient in the miR-146a gene have chronic myelproliferation in bone marrow and spleen concurrent with increased NF-κB activation. 56 Using this model, they observed massive accumulation of CD11b+ Gr1+ myeloid progenitors (i.e. MDSCs) in spleen and lymph node upon challenge with a lethal dose of bacterial LPS. 55 miR-146a may mediate this effect by targeting M-CSFR, as its level was increased in the absence of miR-146a. Given that miR-146a negatively regulates the signaling proteins IRAK1 and TRAF6, which mediate NF-κB activation in response to inflammatory signals, it was concluded that miR-146a may provide a molecular brake on inflammation. 55 In line with this, miR-146a-deficient mice were hypersensitive to bacterial LPS, as demonstrated by overproduction of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6, in response to injection with a sub-lethal dose of LPS. 55 Of note, Gr1+ CD11b+ MDSCs expand under all inflammatory conditions involving STAT3 and NF-κB activation.13,22 In a model of murine sepsis we found that the miR-146a levels in bone marrow cells increased during early sepsis, but returned to normal levels during late sepsis response where MDSCs accumulate in the bone marrow 20 (and unpublished data).
When the effects of miR-146a inhibition were analyzed in the monocytic population in the bone marrow of miR-146a knockout mice, there were increased percentages of myeloid progenitors that were Ly6Chi CD115+ (i.e. expressing M-CSF), 62 a phenotype of MDSCs; 63 however, there were no increases in the more mature Ly6Clo monocytic cells. 62 Thus, miR-146a appears to be selectively up-regulated upon progenitor cell maturation. In addition, LPS challenge increases the percentages of Ly6Chi cells undergoing cell division in the bone marrow. 62 Together, these studies suggest that miR-146a controls the expression of monocytic progenitors in the bone marrow during acute inflammation and that NF-κB activation in the absence of miR-146a increases immature myeloid progenitors. The effect of NF-κB activation on MDSC expansion during murine sepsis requires engagement of MyD88 upstream of NF-κB. 26 We found miR-146a is expressed at baseline levels in the bone marrow concurrent with expansion of MDSCs during murine sepsis 20 (and unpublished observations). More studies on a direct role for miR146a are needed.
miR-21 and miR-181b
miR-21 is expressed in the myeloid lineage cells 64 and induced in macrophages by the NF-κB pathway downstream of MyD88; 65 miR-21 then acts as a negative feedback regulator of NF-kB. During chronic cellular proliferation, miR-21 supports NF-κB signals and promotes inflammation. 66 miR-21 expression is controlled by the IL-6-STAT3 pathway during epithelial cell proliferation 67 —a pathway essential for expansion and suppressive activity of MDSCs.13,22 Induction of miR-21 together with miR-181b by the inflammatory cytokine IL-6 inhibits tumor suppressor proteins PTEN and CYLD, increases Akt activity and activates NF-κB, which is required for cellular proliferation and maintaining the transformed state in human epithelial cell lines. 67 In this work, it was concluded that miR-21 is part of a positive feedback loop that generates an epigenetic switch that links inflammation to cancer. 67 Thus, miR-21 can both positively and negatively control the NF-κB pathway, depending on the cell type.
Recent findings show that the zinc finger protein, growth factor independent-1 (GFI-1), binds to and represses the miR-21 promoter in mouse and human myeloid cells. 59 GFI-1 is a transcription repressor required for normal granulopoiesis 59 and is rapidly induced by cytokines that control innate and adaptive immune responses; it represses genes implicated in cell proliferation and survival, and promotes terminal differentiation of the mouse and human myeloid progenitor cells. 68 Overexpression of miR-21, together with miR-196b, in mouse bone marrow multipotent progenitor cells in vitro blocks granulopoiesis in response to G-CSF, a phenotype also seen in GFI-1 deficient mice. 59 Under physiological conditions, GFI-1 expression is induced during the transition from the common myeloid progenitor to the granulocyte-macrophage progenitor stage, 69 concomitant with reduced miR-21 expression. 59 This pattern leads to differentiation of granulocyte progenitor cells. Overexpression of miR-21 in mouse bone marrow progenitors also expands the monocytic progenitor population and decreases granulocytic progenitors in the colony-forming assay in response to G-CSF stimulation, whereas miR-196b has the opposite effect. 59 These studies show that deregulated expression of miR-21, in conjunction with other miRNAs, can induce changes in the myeloid proliferation and differentiation program, and influence the myeloid lineage cells, perhaps influencing MDSC expansion.
Like miR-21, miR-181b is a downstream target of STAT3 activation. 67 miR-181b inhibits the CYLD (an inhibitor of NF-κB) and, along with miR-21, activates NF-κB to support epithelial cell proliferation and survival. 67 miR-181b also inhibits another tumor suppressor gene, metalloprotease inhibitor TIMP3, which can transform cells. 70 Elevated levels of miR-181b have been observed in human promyelocytic leukemia, with accumulation of myeloid progenitors. 71
STAT3 is a major mediator of many inflammatory cytokines and growth factors, including IL-6, IL-10 and vascular endothelial growth factor, 72 all of which are implicated in MDSC expansion and immunosuppressive activity.13,22 Recently, Zhang et al. 73 reported that STAT3 was necessary for emergency myelopoiesis and expansion of myeloid progenitors in mouse bone marrow following administration of G-CSF, whose level increases dramatically during infection in mice. 74 STAT3 activates miR-21 and miR-181b expression downstream of IL-6 in human epithelial cells, 67 and also induces expression of the transcription factor C/EBPβ, which promotes emergency myelopoiesis and expansion of MDSCs in mice in response to G-CSF or G-CSF.75,76 Importantly, mice with conditional deletion of STAT3 in the bone marrow compartment do not exhibit emergency myelopoiesis in response to G-CSF administration or bacterial infection, i.e. they had decreases in immature myeloid progenitors. 73 Thus, combined activation of STAT3 and NF-κB expands MDSCs. 22 In support of this, Delano et al. 26 showed that mice deficient in MyD88, an upstream effector of NF-κB activation, do not accumulate MDSCs in the bone marrow or secondary lymphoid organs, such as spleen, during sepsis, similar to wild type mice. This supports the hypothesis that upon STAT3 activation expression of miR-21 and miR-181b can activate NF-κB and dysregulate myelopoiesis, and expand MDSCs during infection or cancer. Preliminary work indicates that both miR-21 and miR-181b expression is modified in mouse bone marrow during the late inflammatory response of sepsis (unpublished data), which is associated with massive expansion of bone marrow MDSCs. 20 More work is needed to determine the functional properties of these putative MDSCs.
miR-155
miR-155 positively and negatively regulates innate and adaptive immune responses,47,77 but its role in myeloid development is less clear. miR-155 is expressed at relatively high basal levels in immature hematopoietic cells under steady-state conditions compared with bone marrow mature lineages. 54 During inflammatory responses, miR-155 levels are further elevated in immature hematopoietic cells and at the same time is induced in developing myeloid progenitor cells to augment production of mature monocytes and dendritic cells. 54 Enforced expression of miR-155 in the mouse bone marrow produces a myeloproliferative phenotype similar to that seen after LPS injection,58,78 and this may be due to miR-155 down-regulating transcription factors known to regulate myeloid biology, including PU.1 and C/EBPβ·54,78 Sustained expression of miR-155 in mice under inflammatory stress increases the granulocytic lineage cells, and this may be due to miR-155 inhibiting inositol-5-phosphatase 1, thus repressing PI3K and the downstream Akt-NF-kB pathways.54,78,79 In addition, dysregulation of miR-155 expression has been linked to cancer and autoimmunity. 54 Mice overexpressing miR-155 develop myeloproliferative disorder similar to certain types of AML in patients with accumulation of myeloid blast cells, which often follows overproduction of myeloid cells under physiological stress.54,78,80 Importantly, miR-155 is strongly induced during emergency myelopoiesis associated with systemic inflammation. 78 Emergency myelopoiesis may compensate for loss of innate immune cells during the early response to acute inflammation or during tumor growth. 19 These studies demonstrate that dysregulated miR-155 expression affects development and function of myeloid lineage cells under inflammatory conditions. We find elevated levels of miR-155 in mouse bone marrow MDSCs during acute sepsis and emergency myelopoiesis (unpublished observations).
miR-17-5p, miR-20a and miR-106a
miR-17-5p and miR-20a belong to the miR-17-92 cluster located on human chromosome 13, whereas miR-106a is related to the paralog cluster miR-106a-363 located on chromosome X.81,82 miR-17-5p, miR-20a and miR-106a influence myeloid development, as evidenced by decreased expression during differentiation of human hematopoietic CD34+ progenitor cells into monocytes when stimulated in vitro with M-CSF. 11 These decreases in miRNA levels correlate with de-repression of the transcription factor AML1, also known as runt-related transcription factor 1. The resulting increase in AML1 promotes human monocyte differentiation and maturation. In contrast, overexpression of miR-17-5p, miR-20a and miR-106a in human CD34+ progenitor cells represses AML1 by binding to its promoter, which results in the down-regulation of M-CSFR, thus limiting myeloid differentiation. 11 Because AML1 also increases M-CSFR expression, 11 decreased expression of these three miRNAs during myelopoiesis may provide a negative feedback loop, wherein AML1 and M-CSFR expression is increased, and the miRNA expression is decreased. Because repression of AML1 promotes accumulation of myeloid progenitor blasts and AML, 9 aberrant expression of these three miRNAs could contribute to MDSC accumulation. Indeed, elevated expression of miRs-17-5p, 20a and 106a in MDSCs from bone marrows of mice with sepsis has been found (unpublished observations). In addition, a recent study implicated miR-17-5p and miR-20a in regulating the immunosuppressive activity of MDSCs. 83 Transfection of these two miRNAs in splenic MDSCs from tumor-bearing mice reduced their activity to suppress Ag-specific CD4+ and CD8+ T cells by reducing STAT3 activity and production of reactive oxygen species. 83
miR-424
miR-424 regulates monocytic differentiation in human pro-myelocytic cell lines and CD34+ hematopoietic progenitors by forming a temporal regulatory circuitry with the transcription factors PU.1 and NFI-A. 84 PU.1 is crucial for monocytic differentiation and maturation and, also induces miR-424 expression during 12-O-tetradecanoy1 phorbol 13-acetate mediated monocytic differentiation of human NB4 promyelocytic cells. 84 Under these conditions, PU.1 induced miR-424 to down-regulate the expression of NFI-A, an inhibitor of monocyte differentiation, 84 thereby enhancing M-CSFR expression and monocytic differentiation. 84 NFI-A is also down-regulated by miR-223 to promote differentiation of human pro-myelocytic cells into granulocytes in response to retinoic acid stimulation. 60 Thus, repressing NFI-A affects both monocytic and granulocytic differentiation, depending on whether it interacts with miR-424 or miR-223 respectively. Thus, interaction of miR-424 with myeloid transcription factors could influence myeloid differentiation.
miR-125b
miR-125b belongs to a family that includes miR-125a and miR-125b2.54,85,86 While all miR-125 family members are highly expressed in the nervous system, 86 miR-125a and miR-125b are enriched in the mouse and human hematopoietic compartment. 87 The miR-125 family down-regulates proteins involved in cell development and can expand immature hematopoietic cells,54,87,88 as seen when miR-125b expression is up-regulated in acute and chronic myeloid leukemias.89,90 Also, Chaudhuri et al. 91 reported that overexpression of miR-125b in mouse bone marrow produced a non-leukemic myeloproliferative disorder by targeting the pluripotency factor Lin28.
miR-125b transcription is partially regulated by the Akt-NF-κB-dependent pathway. 92 miR-125b expression is diminished in bone marrow-derived macrophages following LPS stimulation,92,93 and LPS-stimulated Akt-/- macrophages express high levels of miR-125b. 92 Whether miR-125b members play a specific role in generating MDSCs is unknown, but its down-regulation by an inflammatory signal like LPS suggests that it may play a negative role in MDSC expansion during inflammatory responses.
miRNAs directly involved in MDSC expansion
Many or most of the miRNAs in the preceding discussion are candidates for regulating MDSC plasticity, but the data are as yet inconclusive. We consider the role of miR-223 and miR-494 more definitive as MDSC regulators.
miR-223
miR-223 was one of the first miRNAs linked to myeloid development, and its role in myeloid cell proliferation and differentiation has been extensively studied both in vitro and in vivo.12,54,94 miR-223 is expressed in the cells of the myeloid lineage and its levels increase during differentiation and maturation of the granulocytic lineage in mouse bone marrow. 12 Accordingly, it has been postulated that miR-223 negatively regulates myeloid progenitor proliferation and expansion. 12 High levels of miR-223 promote myeloid progenitor differentiation into mature granulocytes12,46 and miR-223-deficient mice expand the granulocyte progenitor compartment owing to the progenitor's inability to terminally differentiate into granulocytes and develop inflammatory lung pathology after systemic bacterial LPS challenge. 12 Thus, miR-223 plays a positive role in granulocytic differentiation and its absence is associated with expansion of immature myeloid progenitors, which can lead to accumulation of MDSCs in the presence of stress signals produced by a growing tumor or an infection.
miR-223 expression is lower in AML, where there is accumulation of myeloid progenitors owing to a block in progenitor differentiation.53,71 miR-223 expression is also decreased in the MDSCs from tumor-bearing animals, whereas its target, the myeloid enhancer factor 2 c (Mef2c), is increased and promotes myeloid progenitor proliferation and survival. 94 In this context, the tumor-associated factor prostaglandin E2 down-regulated miR-223 and increased Mef2c, which promoted MDSC accumulation. 94 Moreover, up-regulation of miR-223 in tumor-bearing animals reduced MDSCs, and inhibiting Mef2c in the presence of miR-223 increased MDSCs. 94
A signaling circuit comprising the CCAAT enhancer-binding protein C/EBPα, the related transcription factor NFI-A and miR-223 itself controls levels of miR-223, 60 where C/EBPα supports miR-223 expression and NFI-A represses it. As a feedback, miR-223 represses NFI-A, thereby activating its own expression. MiR-223 down-regulates the transcription factor Mef2c, which has two conserved miR-223 binding sites in its 3′UTR.12,61 In support of this, Johnnidis et al. 12 have shown that miR-223 represses expression of the luciferase gene containing the Mef2c 3′UTR. Genetic ablation of Mef2c in mouse suppresses myeloid progenitor expansion, whereas miR-223-deficient mice have elevated levels of Mef2c expression and increases in myeloid progenitors in bone marrow. 12 PU.1 also controls miR-223 expression 60 and PU.1 levels increase during granulocytic and monocytic differentiation. 95 Thus, PU.1 and C/EBP proteins are prominent regulators of gene expression in granulocytes and immature monocytes, 96 and transcriptional interaction of these myeloid lineage-specific transcription factors with miR-223 during myeloid differentiation60,97 can contribute to MDSCs expansion under pathophysiological conditions. While C/EBPα is important for generating granulocyte–macrophage progenitors from common myeloid progenitors, as well as induction of miR-223 during granulocytic differentiation under steady-state conditions,60,75 another C/EBP family member (C/EBPβ) is important for inflammatory-induced (or emergency) myelopoiesis,54,73,75 which is associated with massive expansion of MDSCs. 19 In line with this, a recent study has reported rapid generation of MDSCs from hematopoietic precursors of mouse and human bone marrow after stimulation with G-CSF and IL-6. 76 This expansion of MDSCs was entirely dependent on C/EBPβ. While miR-223 expression was not examined under these conditions, it is possible that STAT3 activation downstream of IL-6 reduces miR-223 expression by changing the balance between C/EBPα and C/EBPβ binding activity. STAT3 activation is critical for MDSC expansion13,22 where it enhances C/EBPβ expression and binding to its target. 73 Along this line, a recent study has shown that STAT3 promotes expansion of Gr1+ CD11b+ myeloid progenitors from mouse bone marrow by modulating c-Myc (a cell-cycle accelerator) expression. 73 STAT3 displaced C/EBPα from the c-Myc promoter and induced C/EBPβ binding to the same cognate site in the presence of growth cytokine (G-CSF) or bacterial infection. 73 Given the important role of C/EBPα in inducing miR-223 expression and the notion that both C/EBPα and C/EBPβ compete for the same cognate site at target promoter, it is conceivable, then, that changes in IL-6-STAT3 signaling, such as during inflammation triggered by infection or growing tumor, will reduce miR-223 expression by modulating C/EBPα and C/EBPβ binding at the miR-223 promoter and subsequently promote MDSC expansion.
miR-494
miR-494 is also directly implicated in MDSC expansion. Liu et al. 98 reported that miR-494 is dramatically induced in MDSCs in tumor-bearing mice. Knock-down of miR-494 in vivo reduced MDSC accumulation and suppressive activity, and inhibited tumor growth. In this model, the tumor-derived factor TGF-β1 was solely responsible for miR-494 induction, as treatment of MDSCs with anti-TGF-β1 Ab reduced miR-494 expression. Further mechanistic investigation revealed that miR-494 inhibited the expression of the phosphatase and tensin homolog PTEN and activated the downstream Akt-NF-κB pathway, which enhanced growth/survival signals and thus contributed to MDSC accumulation. 98 As discussed under miR-21 and miR-181b, activation of STAT3 in a variety of proliferating cells induces miR-21 and miR-181b expression and leads to inhibition of PTEN and activation of the Akt-NF-kB pathway, which enhances cell growth and maintains the proliferative state. 67 Given the essential role of STAT3 and NF-κB activation in MDSC expansion and suppressive activity,13,22 it seems that signals that converge on the STAT3 and NF-κB pathways induce miRNAs that can promote MDSC expansion under various conditions.
Concluding remarks
MiRNAs clearly contribute to myelopoiesis under normal and primary myeloid diseases, such as leukemia and myeloproliferation. The observation that a single miRNA can target many genes in different cell lineages, in the same way a single transcription factor does, supports the disparate roles of miRNA in these diseases and in normal myeloid cells.5,54,58 However, less is known about how miRNAs influence MDSC development and functional plasticity—pro-inflammatory or anti-inflammatory, and immunosuppressive—in association with cancer and inflammatory processes that lie outside of the bone marrow. Virtually nothing is known about how these more distal communications take place, although growth factors like M-CSF may contribute. While dysregulated (emergency) myelopoiesis of inflammation informed by growth factors partially may explain the expansion of MDSCs, the molecular nature of the process is unclear. That a large number of miRNAs likely participate in MDSC plasticity, as discussed herein, appears likely, but direct evidence is limited. However, the mechanistic implications of miR-223 and miR-494, and their interplay with transcription factors and specific lineage-specific transcription factors, may inform better understanding of how epigenetic shifts may alter growth, differentiation and functional phenotypes of MDSCs. That dysregulated expression of miRNAs interact with transcription factors, cofactors and chromatin modifiers 99 supports that targeting specific miRNA-regulated pathways may provide novel ways to treat immunosuppressive or hyperactive phases of diseases associated with MDSC accumulation, including cancer and/or inflammation.
Footnotes
Funding
This work was supported in part by National Institutes of Health Grant R15 GM100322 and by funding from East Tennessee State University College of Medicine.
Acknowledgements
I thank Dr Charles McCall (Wake Forest University School of Medicine) for critical discussion.
Conflict of interest
The author does not have any potential conflicts of interest to declare.