
Abstract
We were able to demonstrate reversible, specific and high-affinity binding of radioactively-labelled TGF-β1 (125I-TGF-β1) to immobilized surfactant protein A (SP-A), with an apparent dissociation constant of 53 picomolar at ∼21℃. Addition of a 200-fold molar excess of the latency associated peptide (LAP) prevented and dissociated the binding of 125I-TGF-β1 to SP-A, whereas latent TGF-β1 had no effect. Using a bioassay for TGF-β1 activity—a luciferase reporter assay—we were able to show that SP-A in the presence of TGF-β1 stimulated the TGF-β1 pathway, whereas SP-A alone had no effect. Studies with structural analogues of the distinct SP-A tail domain and head domain indicated that stimulatory activity of SP-A resided in the head domain. No activation of latent TGF-β1 by SP-A was observed. In addition, we observed that SP-A inhibited TGF-β1 inactivation by LAP. These results indicate that SP-A may have a regulatory role in the TGF-β1-mediated processes in the lung.
Keywords
Introduction
The respiratory system makes up a large surface area exposed to the external environment and, consequently, is continuously exposed to pathogens and other airborne particles. Maintaining immunological homeostasis under these circumstances is only partially understood. However, tight regulation of the immune responses in this environment is inevitably pivotal to ensure a suppressed state under ‘normal’ circumstances, while ensuring a robust response when needed. Numerous debilitating, and sometimes lethal, disorders are known, which can be linked to a poor regulation of this delicate immunological balance. On one side there is an increased risk for viral, bacterial and fungal infection, while on the other there are pulmonary disorders, such as bronchopulmonary dysplasia, sarcoidosis, asthma, chronic obstructive pulmonary disease, bronchitis and lung fibrosis.
One of the key regulators of both the innate and adaptive immune responses in the respiratory system is surfactant protein A (SP-A).1–7 It forms the most prominent protein component of pulmonary surfactant and is predominantly expressed in the respiratory system. Nonetheless, SP-A is also present in a variety of other tissues (e.g. intestine, thymus, prostate, mesentery, middle ear, cornea, lacrimal gland, colon and salivary gland), 8 most of which are exposed to the external environment, emphasizing its role in pathogen-rich surroundings. This multimeric protein belongs to the family of collectins, which is best recognized for its function in innate immunity. 8 This function involves both the lectin head and collagenous tail domain of these proteins. The lectin domain recognizes, binds and opsonizes pathogens, while the collagenous domain is involved in phagocytosis and the production of pro-inflammatory cytokines. 8 However, SP-A has also been shown to possess anti-inflammatory properties when its carbohydrate recognition domain is available for receptor binding, suggesting a dual role for SP-A in innate immune responses and thereby possibly being an important modulator of the pro-/anti-inflammatory balance in the respiratory system. 9 This hypothesis is strengthened by the fact that, besides these regulatory mechanisms elicited by multiple receptors for the various SP-A domains, several proteins (e.g. TGF-β1, surfactant protein SP-D and complement component C1q) have been identified which may assist SP-A in tightly regulating the immune responses.10,11 Additionally, several studies have reported that SP-A is involved in the regulation of the complement system by binding to C1q.11–13
Recently, we were able to demonstrate that another potent immunomodulatory and ubiquitously expressed cytokine was shown to associate with SP-A. 14 This cytokine, TGF-β1, is involved in a multitude of crucial cellular processes, such as migration, proliferation, differentiation and apoptosis. TGF-β1 is a pleiotropic cytokine that is expressed by almost all cells and tissues in the human body. TGF-β1 is synthesized and secreted as an inactive molecule, latent TGF-β1, which consists of TGF-β1 non-covalently associated with the latency associated peptide (LAP). Upon activation by heat, acid treatment, proteolytic cleavage and integrins,15–18 TGF-β1 can bind to its specific receptors on the cell surface. The SMAD-dependent signalling cascade is initiated by TGF-β binding, and stabilizing the complex of two type II receptors (TβRII) and two type I receptors (TβRI), which allows the TβRII receptors to activate TβRI by phosphorylating them. These activated TβRI receptors then phosphorylate SMAD2 and SMAD3, which consecutively form trimers with SMAD4. This complex is shuttled to the nucleus where it regulates the transcription of target genes [e.g. plasminogen activator inhibitor 1 (PAI-1)] by binding co-activators/repressors and subsequent interaction with SMAD-binding elements in target gene promoters. An interaction between SP-A and TGF-β could have important consequences for the involvement of SP-A in numerous processes in the lung, such as inflammation, repair and development.
Therefore, in the current study, we investigated the binding of TGF-β1 to SP-A using immobilized SP-A and radioactively-labelled TGF-β1, and demonstrate that SP-A specifically binds TGF-β1 with high affinity. Additionally, we show that complex formation has biological implications by studying TGF-β pathway activation by the SP-A/TGF-β complex in the well-established PAI/L assay. Using the same assay, the activation of latent TGF-β by SP-A was studied.
Materials and methods
Proteins
Recombinant human hydroxyproline deficient SP-A (rSP-Ahyp) was a kind gift from Dr W. Steinhilber, Nycomed (Constance, Germany). Human TGF-β1, recombinant human LAP, recombinant human latent TGF-β1, and human TGF-β1 and TGF-β2 ELISA kits were obtained from R&D Systems (Minneapolis, MI, USA). 125I-TGF-β1 was obtained from Perkin Elmer (Waltham, MA, USA), human C1q from Calbiochem (La Jolla, CA, USA) and the BCA protein assay from Thermo Fisher Scientific (Rockford, IL, USA).
Human platelets
Human platelets were isolated from 40 ml human blood as described previously.
19
The cellular pellet was suspended in 4 ml of 10 mM HEPES, 6 mM n-octyl β-
SP-A isolation
Human pulmonary SP-A was isolated from bronchoalveolar lavage material from alveolar proteinosis 20 patients as described previously. 21 After isolation, the protein content was determined by BCA protein assay. Subsequently, SP-A was dialyzed against 50 mM ammonium carbonate and lyophilized.
Preparation of TGF-β free SP-A
TGF-β free SP-A was prepared using deoxycholate as described previously. 14 The protein concentration of the pool was determined by a BCA protein assay, the presence of SP-A by SDS-PAGE, and the amount of TGF-β1 and TGF-β2 after acid treatment using the TGF-β1 and TGF-β2 ELISA.
Coupling of SP-A to beads
SP-A was coupled to 1-1’-carbonyldiimidazole-activated Trisacryl GF-2000 beads (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer's instructions. Briefly, SP-A was incubated with the beads in a 1:1 ratio (5 mg of protein:5 ml of bead suspension) for 2 d in a coupling buffer (40 mM boric acid buffer, pH 9.2) at RT. The remaining binding sites were blocked by incubation in a 100 mM Tris solution at RT. Subsequently, TGF-β associated with the bead-immobilized SP-A was removed by overnight (14 h) incubation in 5 mM sodium deoxycholate (DOC; Sigma-Aldrich, Steinheim, Germany), 0.3 M NaCl, 20 mM boric acid, pH 9.2 (DOC-buffer) in a shaking water bath (37℃). Finally, the beads were suspended in coupling buffer containing 0.02% (w/v) NaN3 and stored at 4℃. The SP-A beads were used within 2 wks for the binding experiments, as longer storage affected the binding properties (data not shown).
Binding assay
The bead suspension (10 µl) was incubated overnight on a rotator (4℃) in 1 ml binding buffer [2.5 mM CaCl2, 150 mM NaCl, 0.1% BSA (w/v), 10 mM HEPES buffer, pH 7.4]. Subsequently, the bead suspension was centrifuged (10,000 g, 20 s), the supernatant discarded and incubated with 125I-TGF-β1 (specific radioactivity: 133 TBq/mmol), and the indicated additions in binding buffer (total 100 µl) for 1 h (unless otherwise indicated) at 37℃, 800 rpm (85 g), in a thermo stated mixer. The beads were then washed three times in 1 ml binding buffer (10,000 g; 20 s), re-suspended in 100 µl binding buffer and analysed using a γ-counter. Specific binding of 125I-TGF-β1 was determined by subtracting the bound radioactively-labeled TGF-β1 in the presence of a 200-fold molar excess of non-labelled TGF-β1 from the bound radioactively-labelled TGF-β1 to the beads. Total non-specific binding of radioactively-labelled TGF-β1 to the beads did not exceed 30% of the total binding of radioactively-labelled TGF-β1 to the beads. For the calculation of the apparent dissociation constant and maximal binding, the one site, total binding accounting for ligand depletion analysis of GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA) was used.
Cell culture
Mink lung epithelial cells (MLECs; which were stably transfected with a construct expressing luciferase under control of a TGF-β-responsive truncated PAI-1 promoter
22
(a kind gift from Prof. Dr D.B. Rifkin, Rifkin Laboratory, Department of Cell Biology, New York University Medical Center), were grown in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FBS (Greiner Bio-One GmbH, Frickenhausen, Germany), 2 mM
TGF-β bio-assay (PAI/L)
On day one MLECs suspended in starvation medium (0.1% FBS) were seeded at 200,000 cells/cm2 in 96-well plates and allowed to attach for 6 h at 37℃ and 5% CO2. Starvation medium was the same as culture medium except for the concentration of FBS, which was 0.1% FBS. Cells were stimulated for 18 h at 37℃ and in 5% CO2 with the indicated agent at the specified concentration in starvation medium. Cells were lysed by addition of 100 µl Steady-Glo Luciferase Assay System (Promega, Madison, WI, USA) to the wells. Well plates were vortexed and the cell suspensions were transferred to an opaque white 96-well plate and analysed on a Spectramax L (Molecular Devices, Sunnyvale, CA, USA) luminescence microplate reader. The microplates were analysed at 25℃ for 1 h in which each well was read 6 times at 570 nm, with a 10 s integration, and the consecutive read-outs were averaged.
Statistical analyses
Statistical analyses of the PAI/L assays were performed in GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA) using the one-way ANOVA with a Bonferroni post-test. P-Values < 0.05 were considered to be statistically significant.
Results
Time course of the binding of 125I-TGFβ1 to SP-A
125I-TGF-β1 was incubated with immobilized SP-A in the presence or absence of a 200-fold molar excess of non-labelled TGF-β1 for the indicated time intervals. It was shown that 125I-TGF-β1 bound specifically to SP-A reaching a plateau after approximately 15 min (Figure 1).
Time course of binding of TGF-β1 to SP-A. Bead-immobilized SP-A was incubated for various time intervals with 2.5 fmol 125I-TGF-β1 in the presence or absence of a 200-fold molar excess of non-labelled TGF-β1. The curve (total minus non-specific binding) shows the specific binding of 125I-TGF-β1 to bead-immobilized SP-A. Each point represents the mean of three observations ± SD.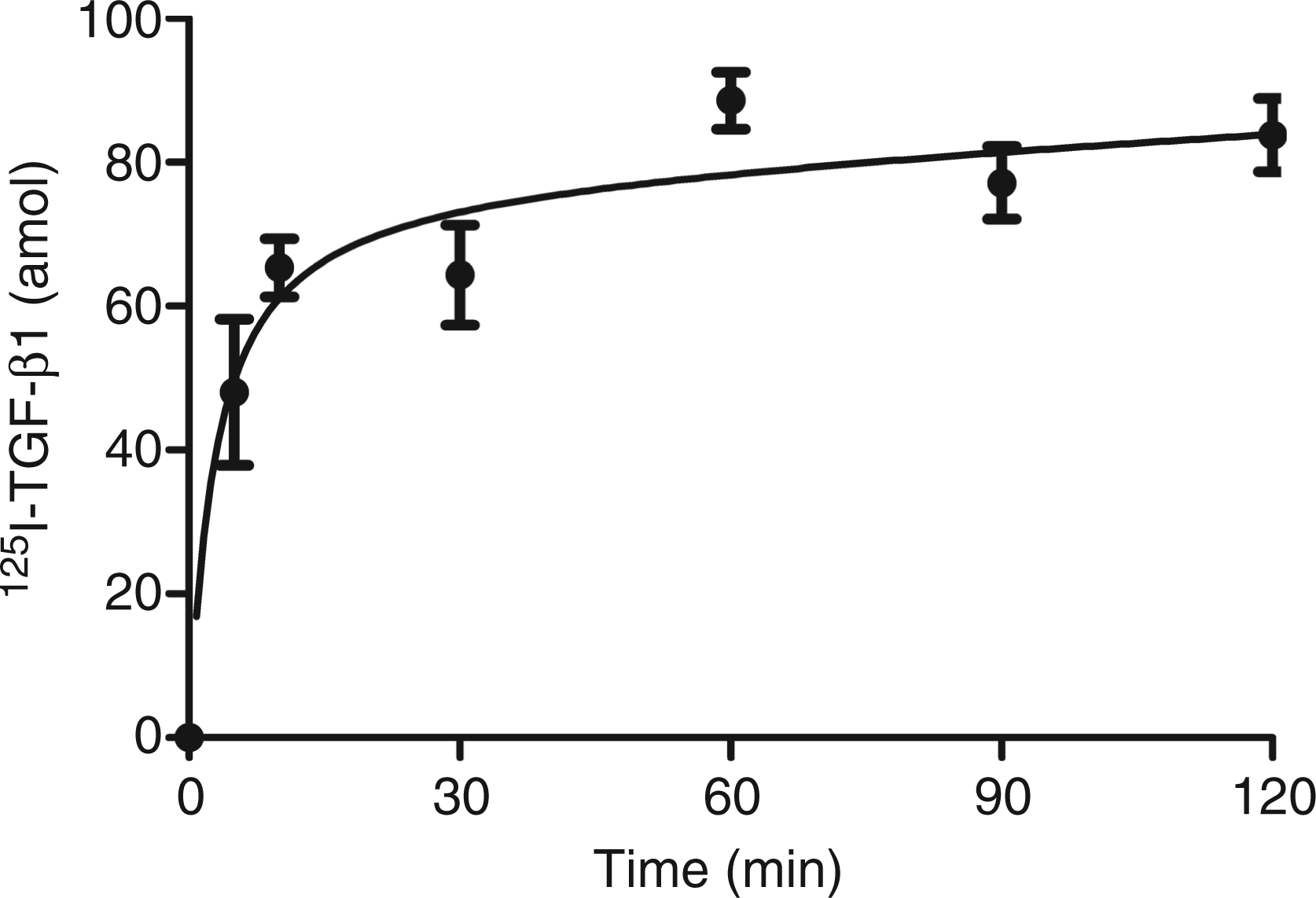
Specificity and reversibility of the binding of 125I-TGFβ1 to SP-A
Increasing concentrations of non-labelled TGF-β1 prevented binding of 125I-TGF-β1 to SP-A (Figure 2A), indicating specific binding of TGF-β1 to SP-A. Latent TGF-β1 did not compete with 125I-TGF-β1 for binding to SP-A. Addition of LAP up to a 10-fold higher concentration than 125I-TGF-β1 had no effect on the binding of 125I-TGF-β1 to SP-A, whereas further increasing the LAP concentration inhibited the 125I-TGF-β1 binding to SP-A (Figure 2A; circles). The reversibility of the binding of 125I-TGF-β1 to SP-A was examined by measuring the dissociation of bound 125I-TGF-β1 when a 200-fold molar excess of non-labelled TGF-β1 was added to the reaction mixture (Figure 2B). Addition of the non-labelled TGF-β1 or LAP, 1 h after the beginning of the incubation, resulted in a dissociation of 125I-TGF-β1 bound to SP-A. This suggests that the binding was at equilibrium and did not become irreversible with time. Latent TGF-β1 could not dissociate bound 125I-TGF-β1 (Figure 2B), which is in line with the previous observation that latent TGF-β1 could not prevent binding of 125I-TGF-β1 to SP-A (Figure 2A).
Specificity and reversibility of the binding of TGF-β1 to SP-A. (A) Bead-immobilized SP-A was incubated for 1 h with 2.5 fmol 125I-TGF-β1 and increasing concentrations of non-labelled TGF-β1(▴), LAP (•) or latent LTGF-β1 (▪). (B) Bead-immobilized SP-A was incubated for 1 h with 125I-TGF-β1. Subsequently, buffer or a 200-fold molar excess of non-labelled TGF-β1, LAP or LTGF-β1 was added to the mixture and incubated for an additional 30 min. Each point or bar represents the mean of three observations ± SD.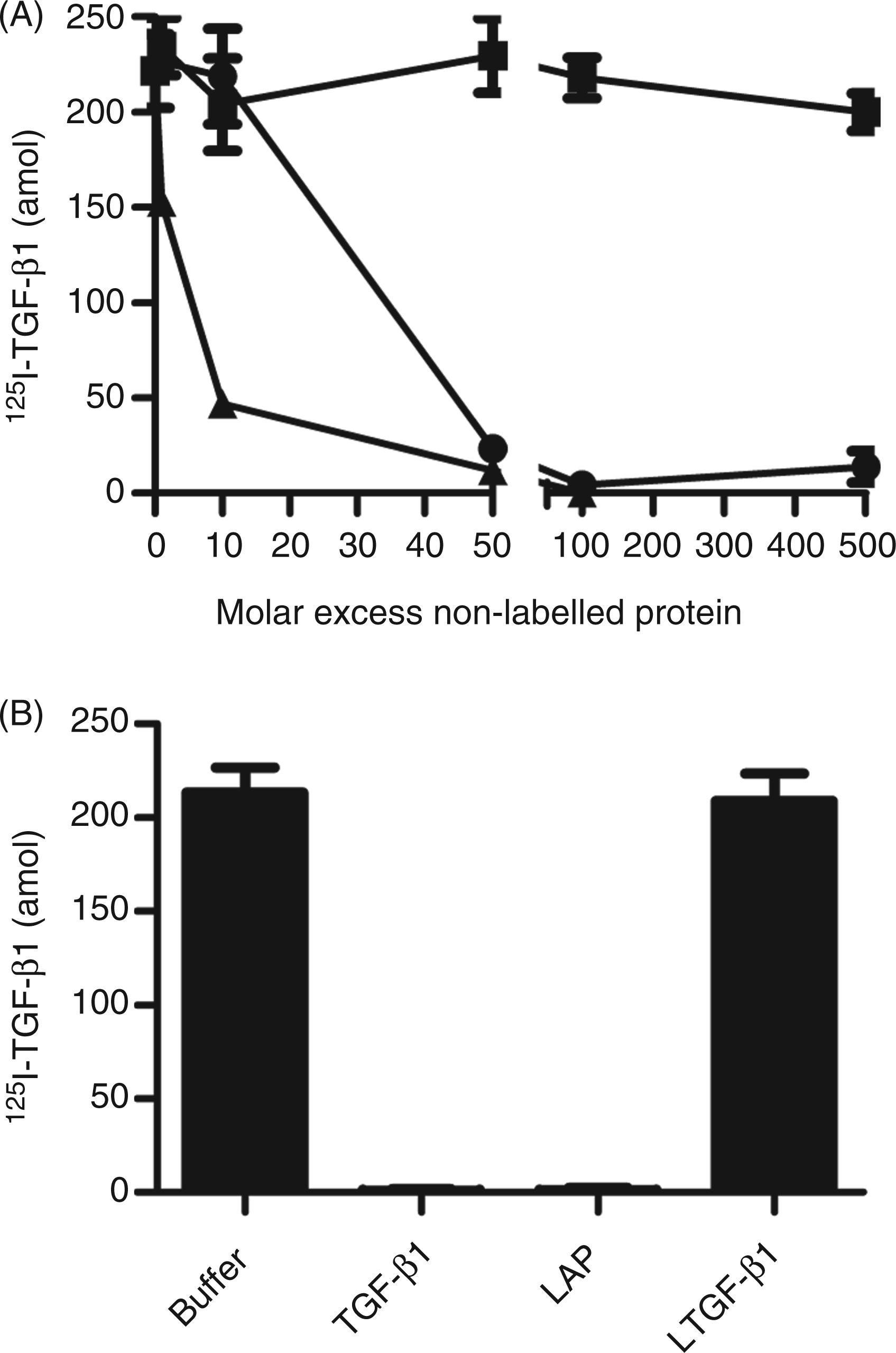
Determination of apparent dissociation constant (Kd) of 125I-TGFβ1 and maximal binding
To determine the affinity of TGF-β1 for SP-A and the maximal amount of TGF-β1 that can bind to SP-A, binding of 125I-TGF-β1 was examined as a function of the TGF-β1 concentration (Figure 3). Analysis of the binding using the one site total binding accounting for ligand depletion analysis yielded an apparent dissociation constant for TGF-β1 of 53 ± 18 pM and a maximal binding of 2.5 ± 0.6 ng TGF-β1 per mg SP-A.
Determination of the apparent dissociation constant. Bead-immobilized SP-A was incubated for 1 h with increasing concentrations of 125I-TGF-β1 and SP-A bound 125I-TGF-β1 was determined. A representative experiment of three experiments is shown.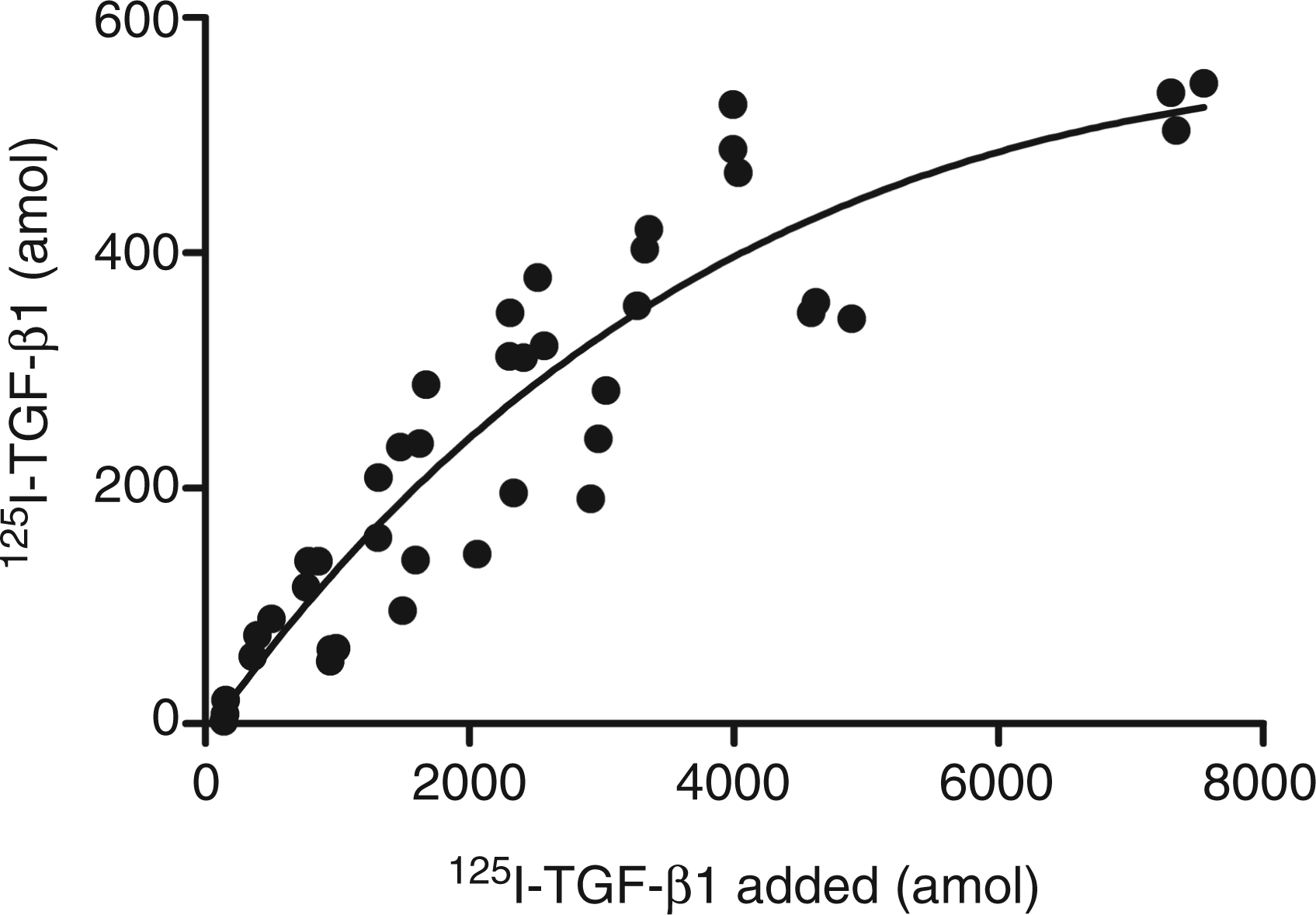
Influence of SP-A on the biological activity of TGF-β1
To study the influence of complex formation of TGF-β1 with SP-A on the biological activity of TGF-β1 we used the PAI/L assay and TGF-β-free SP-A. Increasing SP-A concentrations in the presence of 1 ng/ml TGF-β1 intensified the luciferase expression by 25% (P < 0.01) at 200 µg/ml and 45% (P < 0.001) at 400 µg/ml compared with the control (Figure 4, black bars). This indicates that SP-A enhances the transcription activity of TGF-β1 target genes. No response was observed with SP-A without TGF-β1 in the PAI/L assay (Figure 4, white bars).
Influence of SP-A on the biological activity of TGF-β1. Increasing concentrations of SP-A, pre-incubated for 1 h with 1 ng/ml TGF-β1 (black bars) or without (white bars), were added to MLECs. The cells were incubated for 18 h with the mixtures. Subsequently, the TGF-β pathway activity was assessed by luciferase expression. The data are expressed as mean relative luminescence unit percentage and standard error of three observations compared with control (0 µg/ml SP-A and 1 ng/ml TGF-β1), *P < 0.01, **P < 0.001. A representative experiment of three is shown.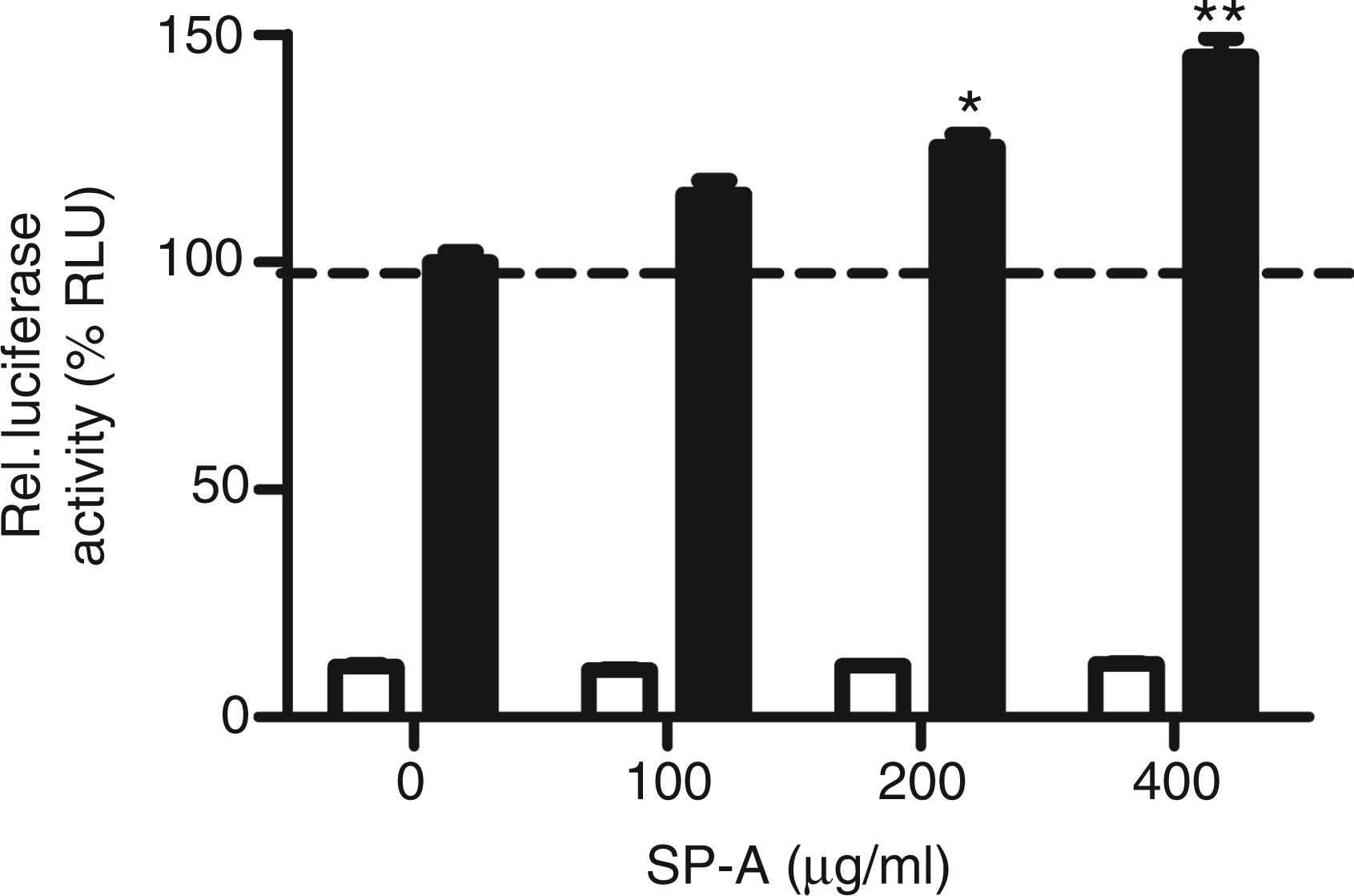
Influence of analogues of functional SP-A domains on the TGF-β1 pathway
In order to establish which part of SP-A, the carbohydrate binding head or the collagenous tail, are responsible for the observed stimulation of the TGF-β1 pathway, we studied the effects of two analogues of these domains. Firstly, a recombinant SP-A analogue deficient in hydroxyproline (rSP-Ahyp), a vital component for the collagenous tail of SP-A, was used as an analogue for the head domain of SP-A.
10
A concentration-dependent increase in relative luminescence compared with control was observed (Figure 5, white bars).
Effects of various SP-A domains on the biological activity of TGF-β1. Increasing concentrations of rSP-Ahyp (white bars) or C1q (black bars) were pre-incubated for 1 h with 1 ng/ml TGF-β1 and added to the MLECS. The cells were incubated for 18 h with the mixtures. Subsequently, the TGF-β pathway activity was assessed by luciferase expression. The data are expressed as mean relative luminescence unit percentage and standard error of three observations compared with control (0 µg/ml rSP-Ahyp/C1q and 1 ng/ml TGF-β1), *P < 0.001. A representative experiment of three is shown.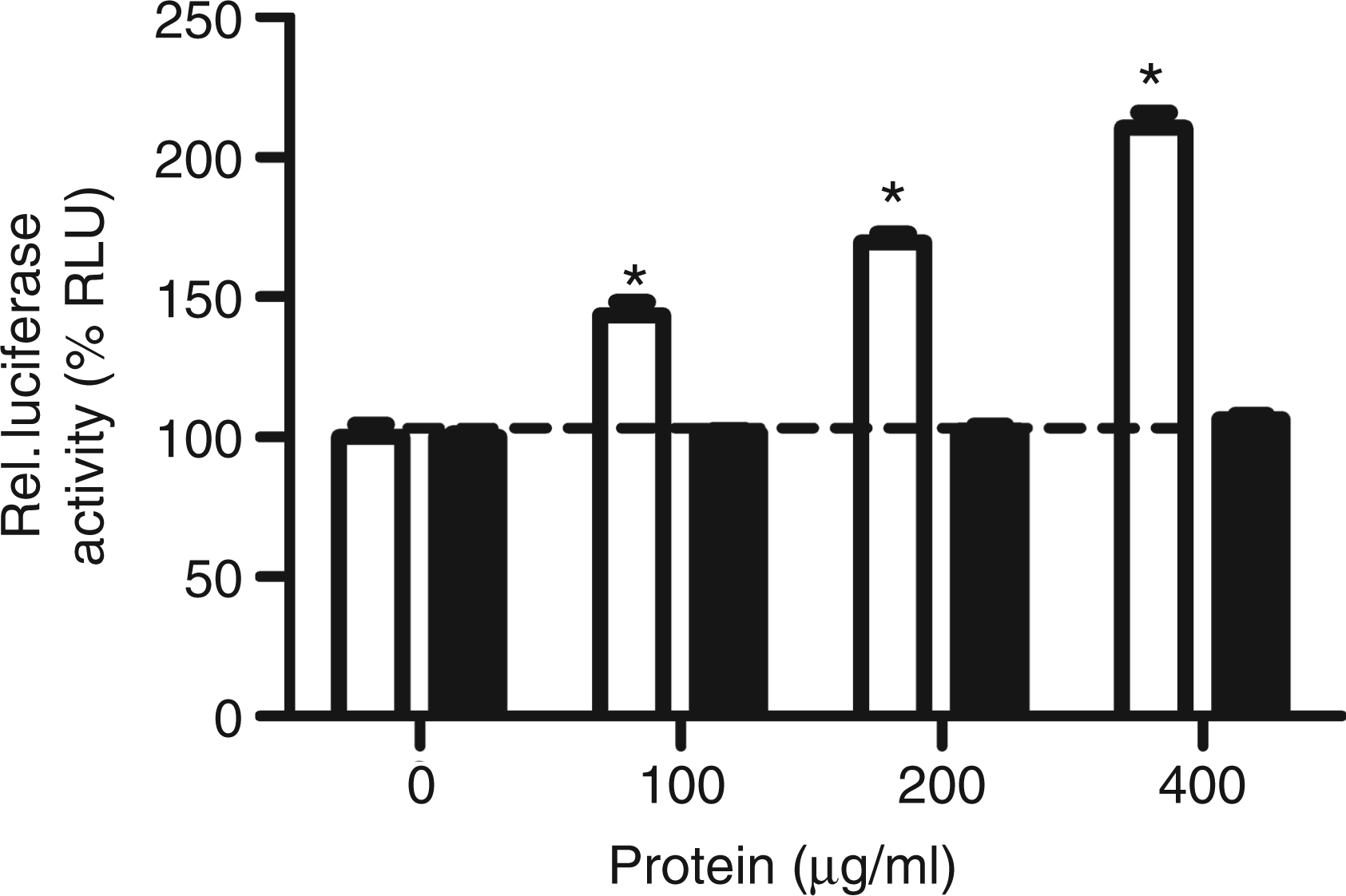
The modulation of the TGF-β pathway by C1q, an analogue of the tail domain of SP-A, 23 was also evaluated. C1q did not significantly alter the luciferase activity (Figure 5, black bars). Neither rSP-Ahyp nor C1q alone changed baseline levels of luciferase activity significantly, indicating that there was no TGF-β present in these preparations (data not shown).
Latent TGF-β1 activation by SP-A
The influence of SP-A on the activation of latent TGF-β was studied using two different sources of latent TGF-β: recombinant human latent TGF-β1 (rlatent TGF-β) and a lysate of human platelets. SP-A had no effect on the activation of either rlatent TGF-β (Figure 6A) or latent TGF-β present in blood platelets (Figure 6B).
Activation of latent TGF-β1 by SP-A. The cells were incubated with increasing concentrations SP-A and either with 8 ng/ml rlatent TGF-β1 (A) or a 10% (v/v) lysate of human platelets (B). The cells were incubated for 18 h with the mixtures. Subsequently, the TGF-β pathway activity was assessed by luciferase expression. The data are expressed as the mean relative luminescence units (RLU) and standard error of three observations. A representative experiment of three is shown. PC: positive control, 8 ng latent acid-treated TGF-β; NC: negative control, starvation medium without additions; PC: positive control, 10% lysate of acid-treated human platelets.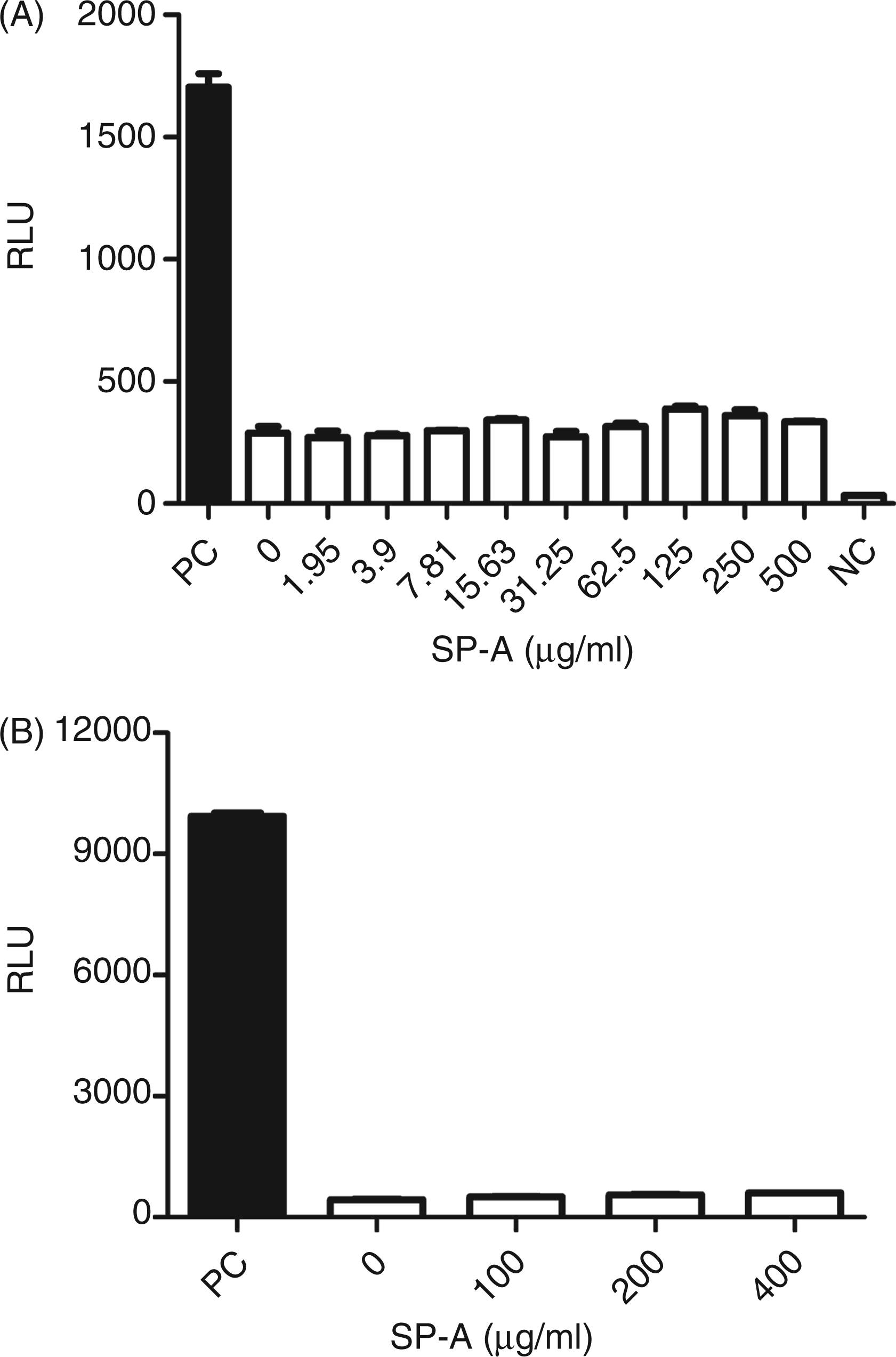
Influence of SP-A on the LAP-mediated inhibition of TGF-β1
LAP inhibits the biological activity of TGF-β1 (Figure 7A). At a LAP concentration of 10 ng/ml, approximately 20% of the added TGF-β1 (1 ng) was inhibited, raising the LAP concentration to 100 ng/ml resulted in a reduction of 85% of the activity of TGF-β1. Addition of SP-A to the incubations reduced the inhibition of TGF-β1 by LAP. This suggests that SP-A limits the inactivation of TGF-β1 by LAP. Similar results were obtained with an acid-treated lysate of human platelets (Figure 7B).
Influence of SP-A on the LAP-mediated inhibition of TGF-β1. The cells were pre-incubated with three different concentrations of SP-A [0 = control (•), 100 µg/ml (▪) and 200 µg/ml (▴)], increasing concentrations of LAP and either 1 ng TGF-β1 (A) or a 10% (v/v) lysate of acid-treated human platelets (B). The cells were incubated for 18 h with the mixtures. Subsequently, the TGF-β pathway activity was assessed by luciferase expression. The data are expressed as the mean activity and SE of three observations as percentage of the incubations which contain no LAP and either 1 ng TGF-β1/ml (A) or 10% acid-treated platelets lysate (B). At LAP concentrations of 10 ng/ml or higher, the values are significant different from the control (no LAP present), P < 0.05. A representative experiment of three is shown.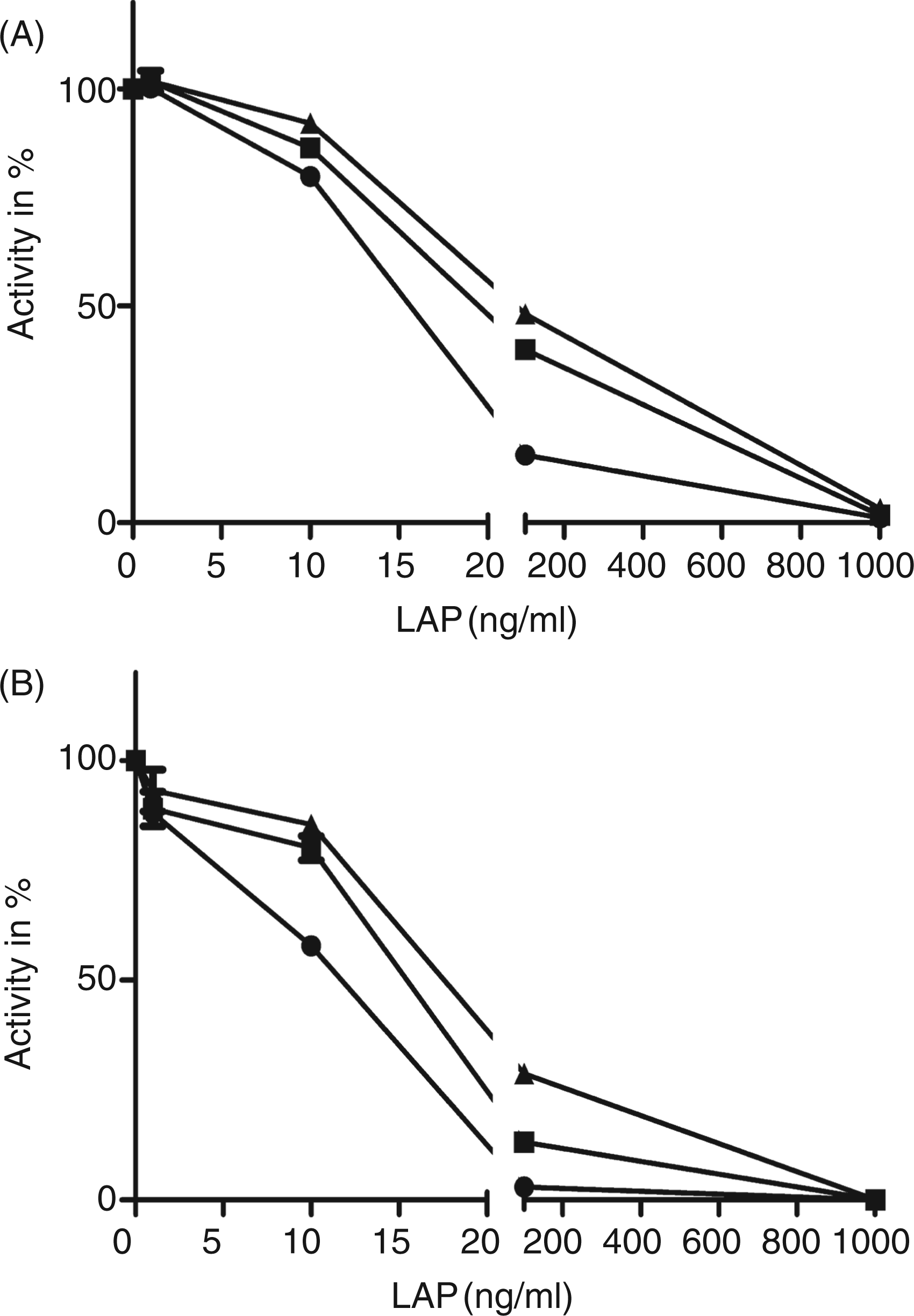
Discussion
In a previous study 14 we were able to demonstrate the presence of TGF-β1 and TGF-β2 in human and porcine pulmonary surfactants, and SP-A purified from these surfactant. The complex of TGF-β1,2 with SP-A was dissociated by deoxycholate treatment or deglycosylation. In the present work, we expanded these studies. We characterized the binding of TGF-β1 to SP-A and determined the effect of association of TGF-β1with SP-A on the biological activity of TGF-β1.
Binding studies with 125I-TGF-β1 and immobilized SP-A indicated that maximal binding of TGF-β1 to SP-A was achieved in 15 min. Increasing the concentration of non-labelled TGF-β1 in the reaction mixture prevented binding of 125I-TGF-β1 to SP-A when it was added at the beginning of the incubation. Furthermore, adding non-labelled TGF-β1 after a 1-h incubation period of SP-A with 125I-TGF-β1, dissociated the latter. These results indicate reversible and specific binding of 125I-TGF-β1 to SP-A. Latent TGF-β1 did not influence the binding of 125I-TGF-β1 to SP-A. This result indicates that TGF-β1, when it is complexed with LAP in the latent TGF-β1 conformation, is no longer able to compete with 125I-TGF-β1 for binding SP-A.
Our data show that LAP inhibited the binding of 125I-TGF-β1 to SP-A in a co-incubation experiment and dissociated SP-A-bound 125I-TGF-β1. This indicates that LAP competes with SP-A for binding TGF-β1. Previous reports have shown that in vitro incubation of LAP and TGF-β1 leads to the formation of a reversible LAP-TGF-β1 complex, with a dissociation constant ranging from 1.1 to 7 nM.24,25 This reported dissociation constant for the LAP TGF-β1 complex is higher than the apparent dissociation constant of 53 pM we established for the SP-A–TGF-β1 complex, which indicates a higher affinity of TGF-β1 for SP-A than LAP. The estimated concentrations of SP-A and TGF-β1 in human alveolar lining fluids are 0.5 mg/ml for SP-A and 6 ng/ml for TGF-β1 respectively.26,27 Assuming that the entire SP-A molecule is in the octadecamer form its MM will be 630 ku and 1 mg of SP-A will be 1.59 nmol. We found that 1 mg of SP-A, 1.59 nmol, will associate with a maximal 2.5 ng of TGF-β1 or 100 fmol (MM TGF-β1 = 25 ku). This indicates that approximately 100,000 molecules of SP-A will contain, maximally, 6.3 molecules of TGF-β1. From the SP-A point of view this is very low. However, a substantial part of the TGF-β1 present in the alveolar lining material will be bound to the SP-A owing to the high affinity of TGF-β1 for SP-A, and maximal binding of 2.5 ng TGF-β1 per mg SP-A. Therefore, complex formation between SP-A and TGF-β1 is relevant for the biological activity of TGF-β1.
We studied the role of SP-A on the biological activity of TGF-β1 using the PAI/L assay. In this assay SP-A, depleted of TGF-β by deoxycholate treatment, 14 had no effect. However, SP-A combined with TGF-β1 enhanced the luciferase activity in a concentration-dependent manner. To determine which part of the SP-A molecule, the lectin head or the collagenous tail domain, is responsible for stimulation, we used analogues of these two domains of SP-A. For the collagenous domain, we used complement factor C1q, as it was demonstrated that collagenous domains of SP-A and C1q are highly similar. Moreover, these proteins share an overall similarity in macromolecular structure and receptor affinity (e.g. calreticulin, CD91, CD93 and integrin α2β1),9,28–37 while they lack sequence homology in the non-collagenous domain. For the head domain we used a recombinant SP-A protein deficient in hydroxyproline (rSP-Ahyp), the critical component of the collagenous domain. Only rSP-Ahyp had a similar stimulatory activity as intact SP-A, suggesting that the lectin head domain of SP-A is responsible for stimulating the TGF-β1 activity.
Having established that SP-A increased the activity of TGF-β1, we wanted to assess whether SP-A influences latent TGF-β1 activation, we incubated SP-A with two different sources of latent TGF-β: recombinant latent TGF-β1 and a lysate of human platelets that contained high concentrations of latent TGF-β associated with the latent TGF-β binding proteins (in order to investigate if SP-A can activate latent TGF-β1). The latter proteins were included as these associated proteins were reported to be essential for some of the activation routes of TGF-β, including integrin-mediated activation.38,39 No activation of latent TGF-β by SP-A was observed when these incubations were tested in the PAI/L assay. A possible explanation for the inability of SP-A to release TGF-β1 from the latent TGF-β1 preparations, as it did for the preparations of LAP and TGF-β1 in the binding assays, is that these complexes have different conformations. Our data indicate that SP-A does not play a role in latent TGF-β1 activation but rather protects uncomplexed TGF-β1 from inactivation by LAP.
Besides inhibiting TGF-β1 inactivation by LAP an effect of SP-A on the TGF-β signal pathway was also observed. SP-A enhanced the TGF-β pathway by elevating the TGF-β dependent expression of luciferase in the PAI/L reporter assay.
In conclusion, we demonstrated specific, reversible and high affinity binding of TGF-β1 to SP-A. Complex formation of TGF-β1 with SP-A inhibited the inactivation of TGF-β1 and stimulated the biological activity of TGF-β1. These results indicate that SP-A influences TGF-β signalling. Further studies are warranted to determine the exact mechanisms of the enhancement of the TGF-β1 signalling pathways to assess the role of SP-A in regulating the immunological balance of the human lung, but also in the multitude of crucial processes (e.g. development and cancer) involving TGF-β signalling.
Footnotes
Funding
This work was supported by ZonMw grant no. 11.400.0083.
Acknowledgements
We thank Dr R. Lutter (Department of Experimental Immunology and Pulmonology Academic Hospital Amsterdam (AMC), the Netherlands) for providing us with alveolar lavage material of alveolar proteinosis patients.