
Abstract
Sporadic inclusion body myositis (sIBM) and polymyositis (PM) are characterized by muscle inflammation, with sIBM showing additional degenerative alterations. In this study we investigated human beta defensins and associated TLRs to elucidate the role of the innate immune system in idiopathic inflammatory myopathies (IIM), and its association with inflammatory and degenerative alterations. Expression levels of human beta-defensin (HBD)-1, HBD-2, HBD-3 and TLR2, 3, 4, 7 and 9 were analysed by quantitative real-time PCR in skeletal muscle tissue. Localization of HBD-3, collagen 6, dystrophin, CD8-positive T-cells, CD-68-positive macrophages, β-amyloid, the autophagy marker LC3, and TLR3 were detected by immunofluorescence and co-localization was quantified. HBD-3 and all TLRs except for TLR9 were overexpressed in both IIM with significant overexpression of TLR3 in sIBM. HBD-3 showed characteristic intracellular accumulations near deposits of β-amyloid, LC3 and TLR3 in sIBM, and was detected in inflammatory infiltrations and macrophages invading necrotic muscle fibres in both IIM. The characteristic intracellular localization of HBD-3 near markers of degeneration and autophagy, and overexpression of endosomal TLR3 in sIBM hint at different pathogenetic mechanisms in sIBM compared with PM. This descriptive study serves as a first approach to the role of the innate immune system in sIBM and PM.
Keywords
Introduction
Idiopathic inflammatory myopathies (IIM) comprise a heterogeneous group of acquired muscular disorders. Sporadic inclusion body myositis (sIBM) and polymyositis (PM) are two of the most common IIM with a distinct phenotype: patients with PM present mostly with proximal muscle weakness, myalgia and high serum-creatine-kinase levels. In the majority of cases, patients with sIBM are of older age and suffer from slowly progressive, often asymmetric, weakness and atrophy of proximal and distal muscles.1,2 Whereas PM is usually responsive to immunosuppressive medication, sIBM is mostly refractory to drug therapy. 3
Endomysial infiltrates of auto-aggressive T-lymphocytes and macrophages, T-lymphocytes invading non-necrotic muscle fibres and muscle fibre necrosis are common histopathological features in sIBM and PM.3,4 At the molecular level, the inflammatory process is characterized by up-regulation of pro-inflammatory molecules, especially of α-chemokines (CXCL-9, CXCL-10), as well as β-chemokines (CCL-2, CCL-3, CCL-4, CCL-19 and CCL-21), and cytokines, including IL-1β and TNF-α.4,5 This pro-inflammatory milieu attracts cytotoxic CD8+ T-cells, which clonally expand and attack myofibres that overexpress MHC I-class molecules. 6
Apart from inflammatory changes, pathological alterations in sIBM include accumulation of aberrant molecules in muscle fibres, especially of β-amyloid. Accumulation of aberrant molecules is associated with proteasome inhibition, endoplasmic reticulum stress and decreased lysosomal degradation.1,4,7 β-amyloid in sIBM muscle is derived mainly from amyloid precursor protein (APP), which has been shown to cause muscle weakness and atrophy in mice. 8 Recent studies have also demonstrated inter-relations of inflammatory processes with degenerative alterations in sIBM, showing that inflammatory mediators like IL-1β co-localize to β-amyloid in myofibres, and that the exposure of human myotubes to IL-1β causes an up-regulation of APP with subsequent accumulation of β-amyloid. 7 Another important aspect of sIBM pathophysiology is the role of autophagic processes and its interrelation with β-amyloid and inflammatory molecules. Skeletal muscle fibres show a high level of constitutive autophagic activity. It has been shown recently that TNF-α, which is overexpressed in muscle tissue of sIBM patients,4,5 leads to an increase of autophagic activity and increased expression of MHC II. 9 Moreover, β-amyloid has been shown to be a substrate of autophagy in sIBM muscle tissue, and is targeted to LC3-positive autophagosomes for further processing. 10 These findings suggest a crucial role of autophagic processes in the pathophysiology of sIBM, which are interrelated with inflammatory and degenerative alterations.
The innate immune system is indispensable for the primary immune defence against invading microorganisms. It comprises cellular and humoral factors, including the ‘host defence peptides’ (HDPs), and human β-defensin (HBD) 1, HBD-2 and HBD-3. They are expressed constitutively in human epithelia or induced after pathogen exposition via TLR-dependent pathways11,12 and are crucial for the primary immune defence against invading pathogens. 13 An illustration of HBD-3 and its association with intracellular signalling pathways is shown in the Supplementary Figure 1.
TLRs recognize different pathogens: TLR2 recognizes lipopeptides/lipoproteins and leads to downstream activation of NF-κB and HBD-3 release. 12 HBD-3 has a bactericidal effect and induces chemotaxis in T-lymphocytes and macrophages.11,14,15 TLR3 binds to host or viral dsRNA and plays a key role in antiviral defence. 16
Apart from the recognition of specific pathogens and initiating host defence mechanisms, the innate immune system is also involved in the development of autoimmune disorders.17,18 Moreover, two recent studies show that the innate immune system also contributes to the pathogenesis of inflammatory myopathies by examination of altered expression patterns for a subset of TLRs in PM and dermatomyositis (DM).19,20
The intention of the study at hand is to give first insights into the role of HBD-3 and TLRs as markers of the innate immune system, and its association with inflammatory, autophagic and degenerative alterations in sIBM and PM.
Material and methods
Patients and muscle biopsies
Patient characteristics; clinical data and biopsy results of all patients.
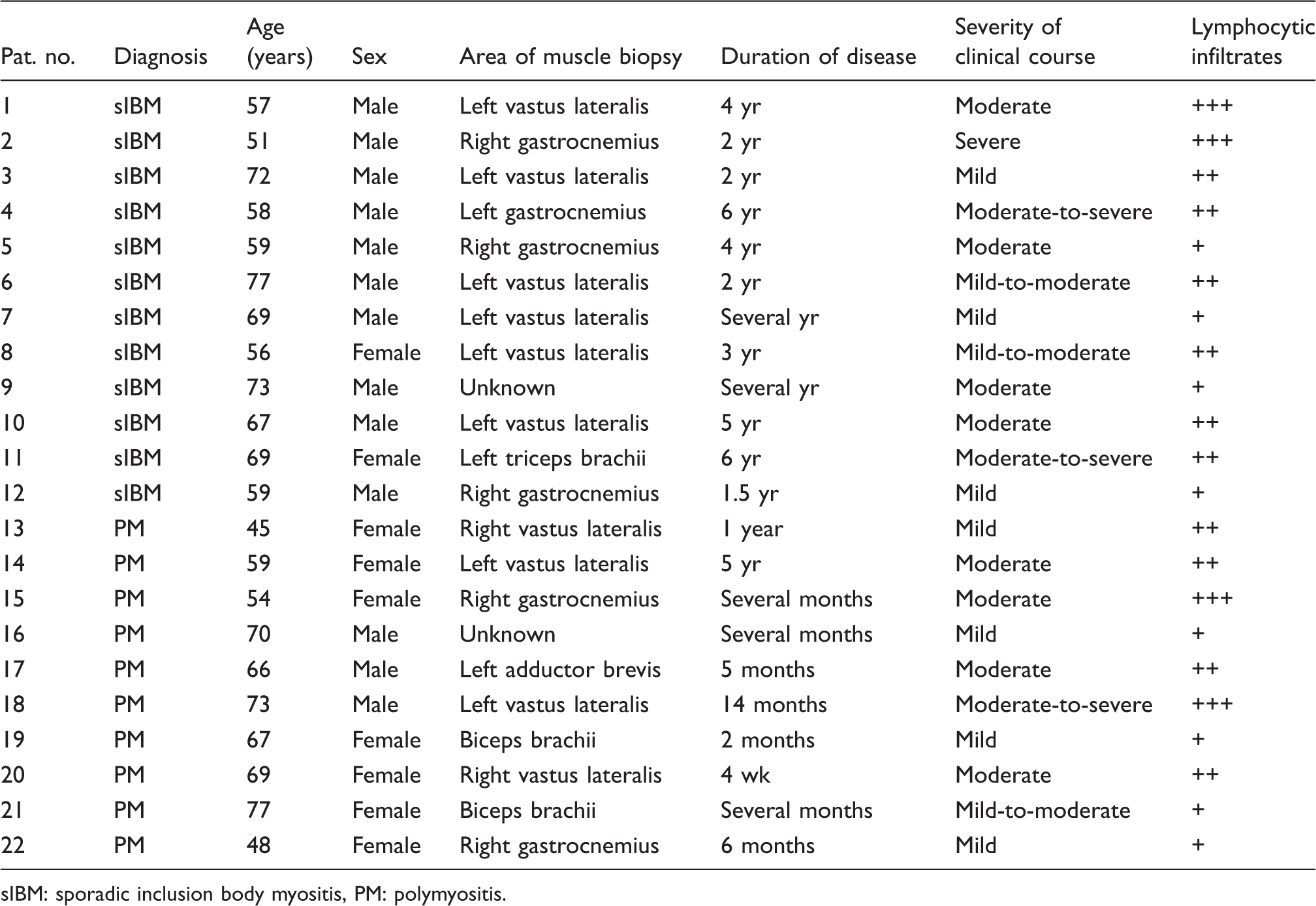
sIBM: sporadic inclusion body myositis, PM: polymyositis.
Isolation of total RNA from tissue
Tissue was stored at −80℃ until further processing. Isolation of total RNA was done using the RNeasy Mini Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions for isolation of total RNA from muscle, including proteinase K pretreatment and on-column DNA-digestion using the RNase free DNase Set (Qiagen). RNA was eluted in a final volume of 30 µl RNase-free water. The concentration of RNA was determined using a biophotometer (Eppendorf, Hamburg, Germany).
Reverse transcription
For reverse transcription, 1 µg of total RNA was used. Total RNA was transcribed into cDNA using the SuperScript™ First Strand Synthesis System for RT-PCR (Invitrogen, Karlsruhe, Germany), following the manufacturer’s instructions for first-strand synthesis using random primers.
qPCR
Overview of primers used for qPCR.

Immunofluorescence analyses
Immunofluoresence studies were performed on 10-µm transverse serial sections of muscle biopsies (specified above). Sections were pre-incubated with BSA (Roth, Karlsruhe, Germany) for 30 min at room temperature (21℃ ± 2℃) to block unspecific binding. Double immunofluorescence stainings were performed using a rabbit polyclonal anti-HBD-3 Ab (Novus Biologicals, Littleton, CO, USA; dilution 1:100), combined with one of the following Abs: mouse monoclonal anti-CD8 Ab (Dako Cytomation, Glostrup, Denmark; dilution 1:100), mouse monoclonal anti-CD68 Ab (Dako Cytomation; dilution 1:200), mouse monoclonal anti-LC3 Ab (Medical and Biological Laboratories, Nagoya, Japan; dilution 1:100), mouse monoclonal anti-β-amyloid (6E10) Ab (Covance, Princeton, NJ, USA; dilution 1:500), mouse monoclonal anti-TLR3 Ab (Abnova, Taipeh, Taiwan; dilution 1:100), mouse monoclonal anti-Dystrophin 3 Ab (Novocastra, Berlin, Germany; dilution 1:40) or mouse monoclonal anti-Collagen 6-Ab (Hybridoma Bank, Iowa City, IA, USA; 1:500). Primary Abs were incubated overnight (16 h ± 2 h) at 4℃. Isotype-specific secondary Abs conjugated with DyLight 488, Cy3 (Dianova, Hamburg, Germany), tetramethylrhodamine isothiocyanate (Dako Cytomation) or Texas red (Jackson Immuno Research, West Grove, PA, USA) were applied according to the manufacturer’s recommendations. Nuclei were displayed by 4´-6-diamidino-2-phenylindole (DAPI) (Roche Diagnostics, Indianapolis, IN, USA; dilution 1:10000). Unspecific staining was excluded by omission of the primary Ab.
Confocal laser scanning microscopy
Samples were evaluated by confocal laser scanning microscopy using a Zeiss LSM 510 (Zeiss, Jena, Germany) and plan neofluar 20 ×/0.4, zoom level 1, 1024 × 1024 pixels, scan speed 6, average number 4. For imaging, the laser module contains a diode laser (405 nm), an argon laser (488 nm), and a helium neon laser (543 nm). To avoid misinterpretation of autofluorescent material in muscle fibres the detection gain and detection offset of the LSM were optimized for each channel (specimen without primary Abs, no fluorescent signal). Thereafter, these parameters were used for all immunohistochemically-stained samples.
Quantitative analysis of the co-localization of HBD-3 and β-amyloid, LC3, CD8, CD68 and TLR3 was performed using the software CoLocalizer Pro. At least 10 different high-power fields (HPF) were analysed out of tissue sections from three different patients to determine correlation of the corresponding co-localization. Co-localization was quantified by counting the relative amount of yellow (co-localization), or exclusively red and green (expression of either β-amyloid, LC3, CD8, CD68 and HBD-3) pixels followed by statistical analysis (Student’s t-test).
Results
HBD-3 specific mRNA was significantly overexpressed in sIBM and PM
As a first approach to the involvement of effector molecules of the innate immune system in IIM, expression levels of HDPs were obtained. Mean expression levels of HBD-3 were increased 100-fold in patients with sIBM (P = 0.000004) and PM (P = 8 × 10−9) related to normal controls. Expression of HBD-1 was detected in 16, of HBD-2 in 11 and of HBD-3 in 5 of the control muscle biopsies (n = 20), showing the following mean expression values of transcript amount per transcript amount of 18SrRNA: HBD-1, 0.6615 ± 0.1004 ng/g; HBD-2, 0.3879 ± 0.1661 ng/g; HBD-3, 0.7384 ± 0.3363 ng/g. Mean expression levels of HBD-1 and HBD-2 were not significantly altered in both IIM (Figure 1).
Quantification of HDPs in IIM. This figure displays the mRNA levels of several HDPs that could be determined in muscle tissue of sIBM and PM patients in relation to healthy control participants’ muscle tissue. Expression levels of HBD-3 were increased 100-fold compared with control muscle biopsies. Expression levels of HBD-1 and HBD-2 were not altered significantly. All values are displayed as mean ± SEM and normalized to 18SrRNA.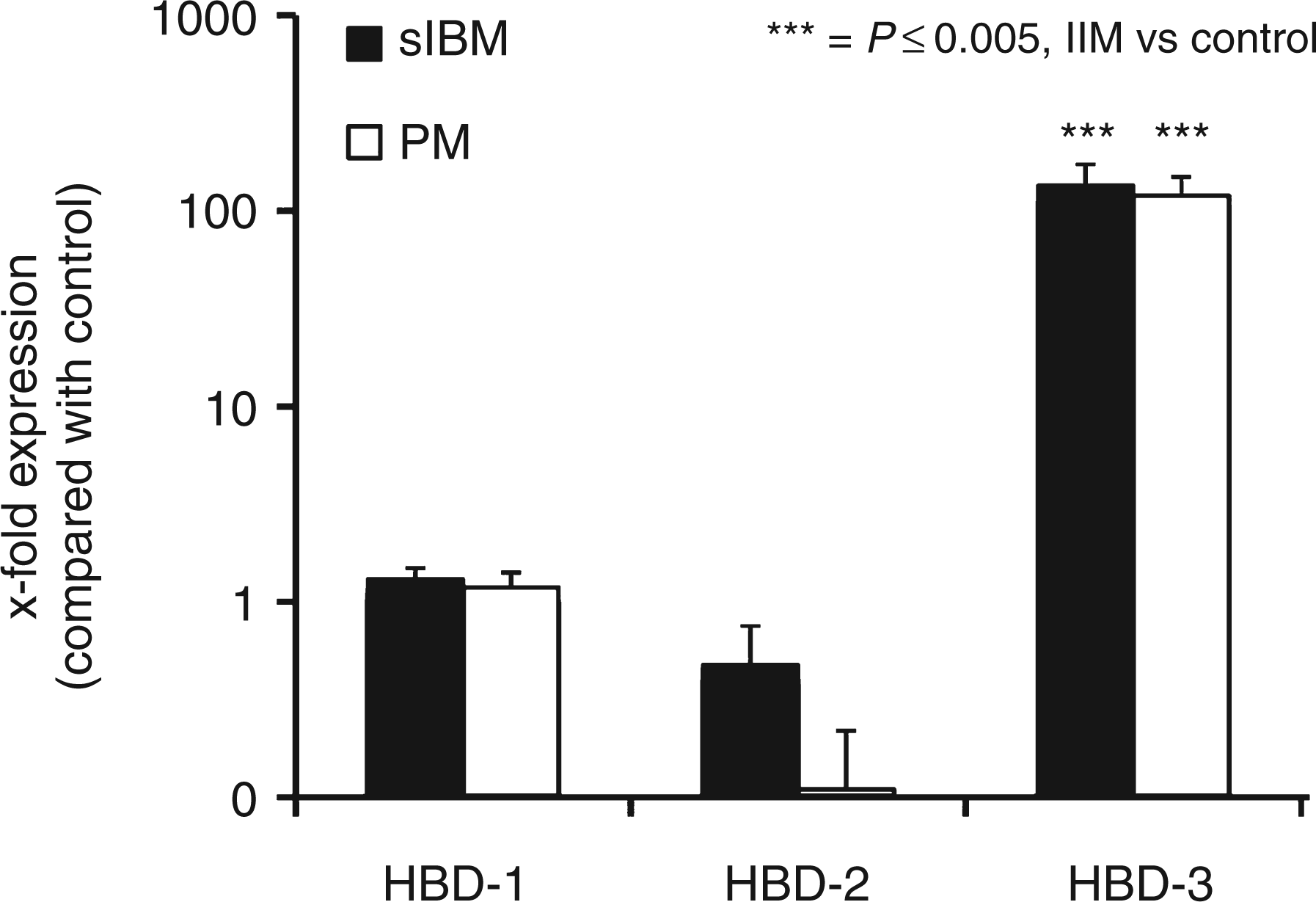
HBD-3 could be located inside muscle fibres and in connective tissue in both IIM, forming ‘aggregate-like’ intracellular structures in sIBM
To obtain the localization of HBD-3 inside muscle fibres and connective tissue, and delineate muscle structure, double-immunofluorescence analyses were performed in cryosections from one sIBM patient, one PM patient and one healthy control participant with HBD-3, Dystrophin 3 and Collagen 6, respectively, as described above. HBD-3 accumulated in an ‘aggregate-like’ manner in sIBM inside Dystrophin 3-positive fibres. It was also diffusely expressed in fibres loosing their dystrophin signal, which we assumed to be pre-necrotic or necrotic (Figure 2A). Figure 2B shows that some fibres in both sIBM and PM muscle tissue expressed HBD-3 diffusely inside the muscle fibre, which may display necrotic or pre-necrotic fibres. Additionally, extracellular expression of HBD-3 was detected in both IIM (Figure 2B).
Localization of HBD-3 in muscle tissue and delineation of muscle structure. The images show HBD-3 (green) and Dystrophin 3 (A, red), as well as Collagen 6 (B, red) double- immunolabelling on fresh-frozen transverse sections of one sIBM patient, one PM patient and one healthy control participant’s muscle biopsy respectively. Nuclei were stained with DAPI (blue). Scale bar = 50 µm.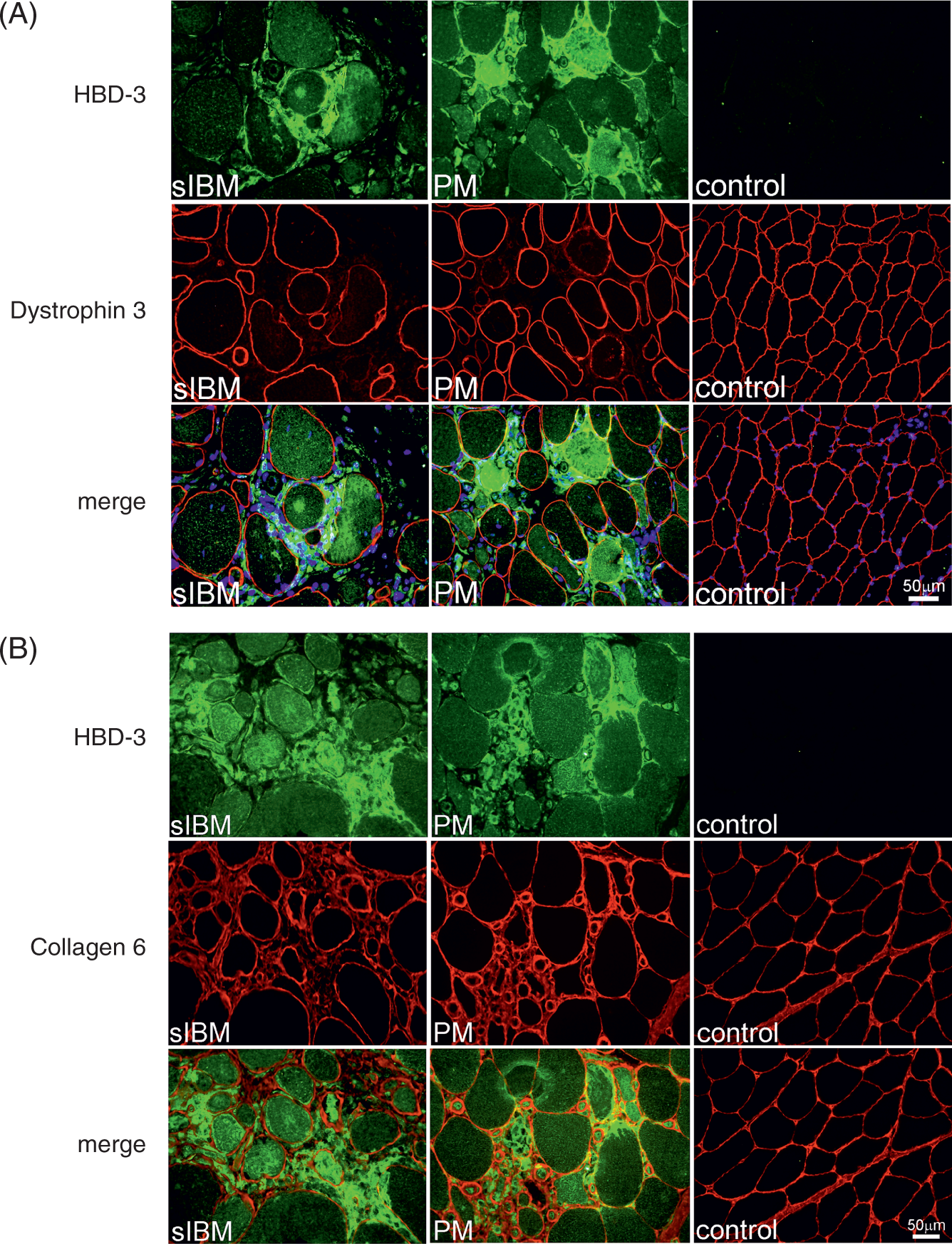
HBD-3 accumulated inside muscle fibres in sIBM and was associated with markers of autophagy, as well as deposits of β-amyloid in sIBM
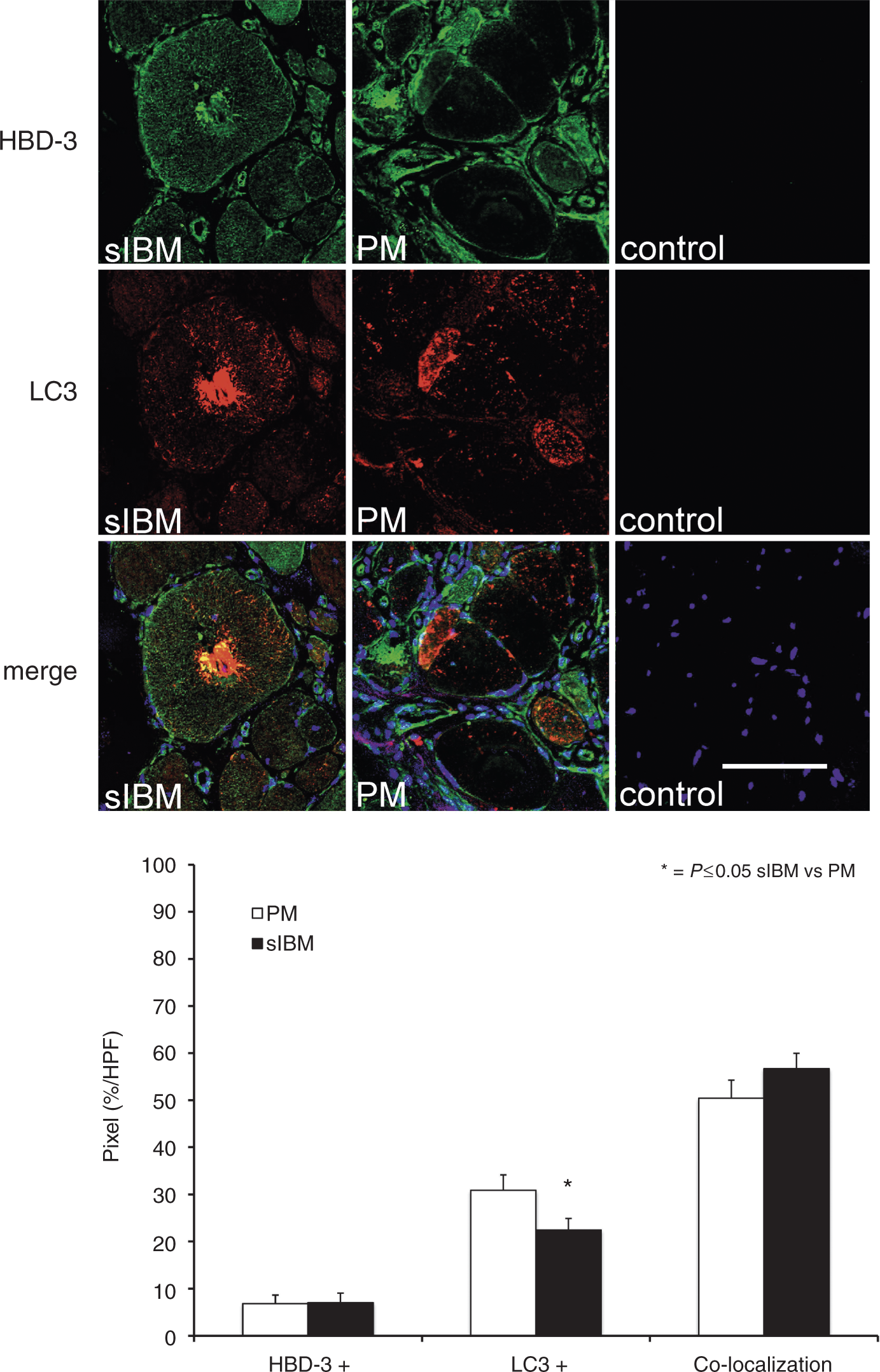
As accumulation of aberrant proteins and autophagic processes are crucial for pathological changes in sIBM, the relationship of HBD-3 to LC3 and β-amyloid was analysed in co-localization studies as described above. HBD-3 co-localized with β-amyloid in muscle fibres from sIBM patients (Figure 3). Serial sections from sIBM muscle tissue stained with HBD-3, β-amyloid, and haematoxylin and eosin (H&E) revealed that HBD-3 was located near vacuolar structures in sIBM. Quantification analysis showed a mean value of co-localization (yellow signal) of 78.77%. Expression of HBD-3 without β-amyloid (green signal) showed a mean value of 1.5% and expression of β-amyloid without HBD-3 (red signal) of 4.5% (Figure 3). These data showed a distinct co-localization of HBD-3 and β-amyloid. To further investigate the association between autophagic activity and the expression of HBD-3, serial sections of sIBM, PM and healthy control muscle tissue were stained with HBD-3 and LC3. HBD-3 co-localized with LC3 in sIBM mainly in fibres with vacuolar structures (Figure 4). Quantification analysis showed a mean value of co-localization (yellow signal) of 56.7% in sIBM and 50.46% in PM (P = 0.22). Expression of HBD-3 without LC3 (green signal) showed a mean value of 7.05% in sIBM and 6.86% in PM (P = 0.95), and expression of LC3 without HBD-3 (red signal) showed a mean value of 22.42% in sIBM and 30.89% in PM (P = 0.04) (Figure 4). The data showed that in sIBM, co-localization of LC3 and HBD-3 was slightly more frequent than in PM.
HBD-3 is located near β-amyloid in sIBM. Immunofluorescence studies for co-localization of HBD-3 and β-amyloid compared with H&E. Fresh-frozen sections of sIBM muscle biopsies (n = 3 patients with each 10 HPF) were double-labelled with HBD-3 (green), β-amyloid (red) and H&E, and analysed by confocal laser scanning microscopy and light-microscopy (H&E). Nuclei were stained with DAPI (blue) in immunofluorescence studies. Co-localization of HBD-3 and β-amyloid is shown in pixels (%/HPF) in sIBM (black bar). Pixels exclusively lightened green or red were shown as HBD-3 + or β-amyloid + respectively. Scale bar = 100 µm. HBD-3 co-localizes with markers of autophagy in sIBM: immunofluorescence studies for co-localization of HBD-3 and LC3. Fresh-frozen sections of sIBM, PM and normal control muscle biopsies (each n = 3 patients with each 10 HPF) were double-labelled with HBD-3 (green) and LC3 (red), and analysed by confocal laser scanning microscopy. Nuclei were stained with DAPI (blue). Co-localization of HBD-3 and LC3 is shown in pixels (%/HPF) in sIBM (black bar) and PM (white bar). Pixels exclusively lightened green or red were shown as HBD-3 + or LC3 + respectively. Scale bar = 100 µm.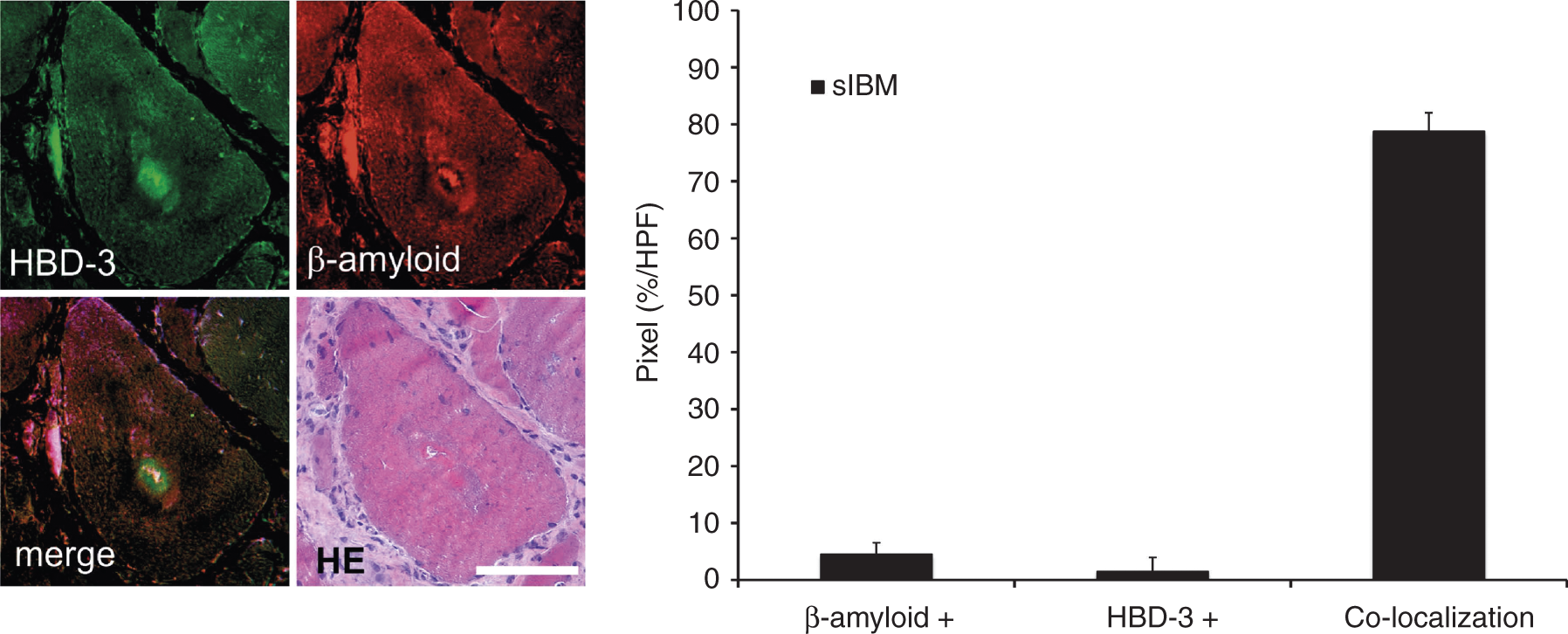
HBD-3 co-localizes with CD8+ T-cells and CD68+ macrophages
To analyse the expression of HBD-3 and its relationship to infiltrating CD8+ T-cells and CD68+ macrophages, co-localization studies were performed. Cryosections of three patients with sIBM and PM each, and two healthy control participants were double-labelled with the aforementioned Abs either against HBD-3 and CD8, or HBD-3 and CD68. Quantification analysis of HBD-3 and CD8 showed a mean value of co-localization (yellow signal) of 56.03% in sIBM and 68.88% in PM (P = 0.001). Expression of HBD-3 without CD8 (green signal) showed a mean value of 18.9% in sIBM and 12.18% in PM (P = 0.03), and expression of CD8 without HBD-3 (red signal) a mean value of 8.32% in sIBM and 9.54% in PM (P = 0.54) (see Figure 5A). HBD-3 co-localized to CD8 in cellular infiltrations in both IIM, showing a significantly (P = 0.014) higher co-localization of CD8 and HBD-3 in PM compared with sIBM (Figure 5A). Quantification analysis of HBD-3 and CD68 showed a mean value of co-localization (yellow signal) of 69.97% in sIBM and 69.16% in PM (P = 0.91). Expression of HBD-3 without CD68 (green signal) showed a mean value of 13.55% in sIBM and 12.62% in PM (P = 0.87), and expression of CD68 without HBD-3 (red signal) showed a mean value of 1.58% in sIBM and 1.62% in PM (P = 0.97) (see Figure 5B). These data showed a distinct co-localization of HBD-3 with CD68+ macrophages invading necrotic fibres in both sIBM and PM patients (Figure 5B).
Co-localization of HBD-3 with CD8 (A) and CD68 (B). Fresh-frozen sections of sIBM, PM and normal control muscle biopsies (each n = 3 patients with each 10 HPF) were double-labelled with HBD-3 (green) and CD8 (A, red) and CD68 (B, red), and analysed by confocal laser scanning microscopy. HBD-3 could be detected in the connective tissue near cellular infiltrations (A) and near CD68+ macrophages (B). Nuclei were stained with DAPI (blue). Co-localization of HBD-3 and CD8, as well as CD68 is shown in pixels (%/HPF) in sIBM (black bar) and PM (white bar). Pixels exclusively lightened green or red were shown as HBD-3 +, while pixels that were only lightened red were displayed as CD8 + (A) or CD68 + (B). Scale bar (A) = 50 µm, scale bar (B) = 100 µm.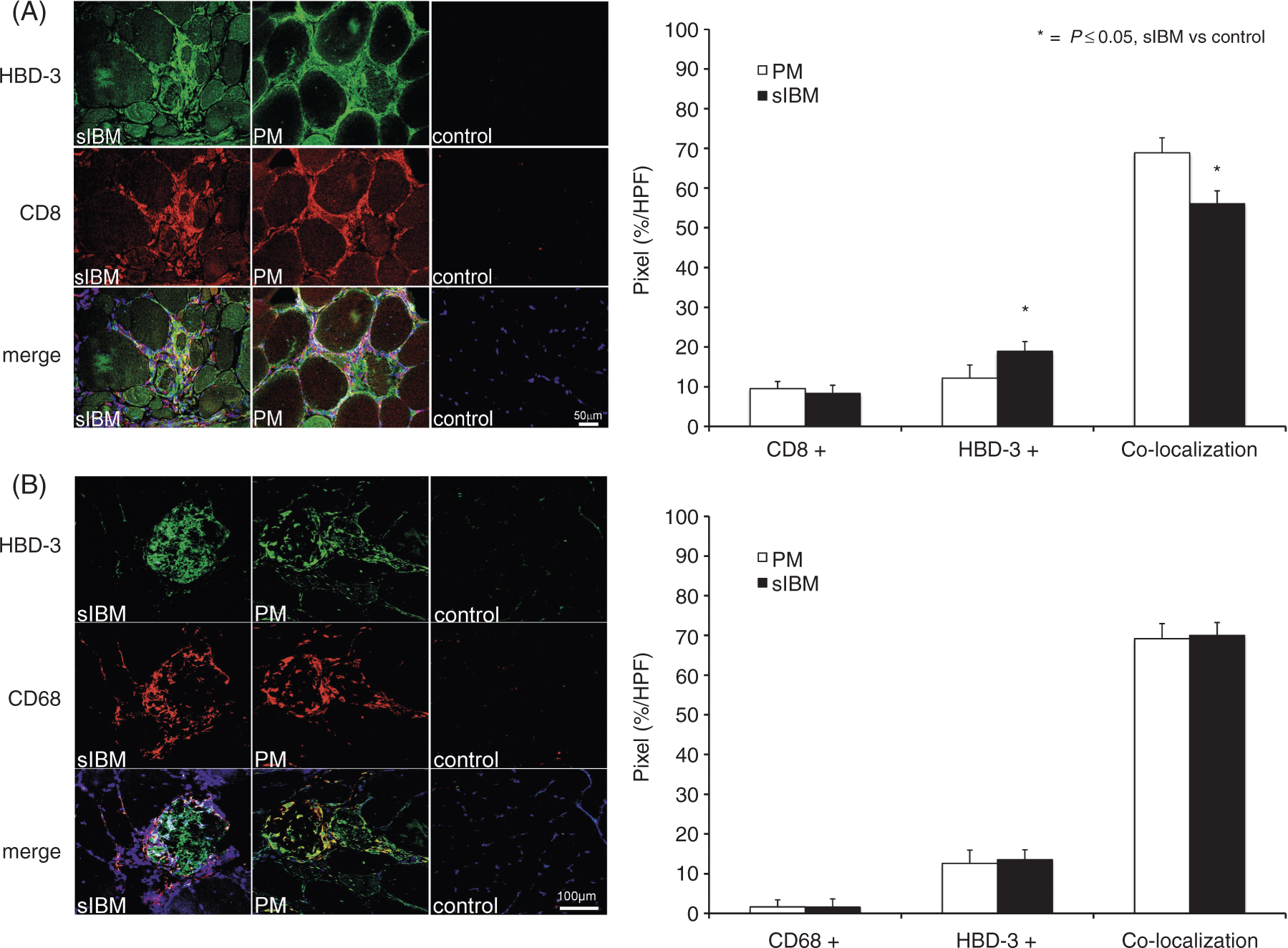
As HBD-3 is mediated by TLR-dependent pathways, specific subsets of TLRs, as well as cytokines and ILs associated with IIM were analysed by qPCR and immunofluorescence.
TLR2, TLR3, TLR4 and TLR7, but not TLR9, were overexpressed in sIBM and PM with increased expression levels of TLR3 in sIBM, and association of TLR3 with HBD-3
Mean expression levels were altered as shown in Figure 6. Expression levels of TLR2, 4 and 7 were increased almost equally in PM and sIBM. The mRNA level for TLR2 was increased 4.5-fold (P = 0.000001) in sIBM and 3.6-fold (P = 0.001) in PM respectively. TLR4 was only slightly increased in sIBM [1.6-fold (P = 0.01)] and PM [1.5-fold (P = 0.008)]. Expression levels of TLR7 were increased the most, i.e. 6.1-fold (P = 0.00015) in sIBM and 5.0-fold (P = 0.00012) in PM. In contrast, expression levels of TLR9 were both significantly decreased in sIBM and PM compared with normal muscle biopsies. Expression levels for TLR3 in sIBM muscle were increased 4.8-fold in sIBM, but only 1.98-fold in PM compared with normal controls (Figure 6). TLR3 was significantly overexpressed in sIBM muscle compared with PM muscle (P = 0.038). Expression of all TLRs was detected in all control samples (n = 20), showing the following mean expression values of transcript amount per transcript amount of 18SrRNA: TLR2, 475 ± 257 ng/g; TLR3, 1460 ± 624 ng/g; TLR4, 4000 ± 1020 ng/g; TLR7, 157 ± 96.4 ng/g; TLR9, 42.2 ± 13.3 ng/g.
Quantitative evaluation of TLRs. Expression levels of TLR2, TLR3, TLR4, TLR7 and TLR9 in sIBM and PM were related to normal controls. Except for TLR9, all TLRs were increased on mRNA level in sIBM and PM compared with normal controls. TLR3 showed a significant higher up-regulation in sIBM (4.8-fold) than in PM (1.98-fold) muscle tissue. All values are displayed as mean ± SEM and normalized to 18SrRNA.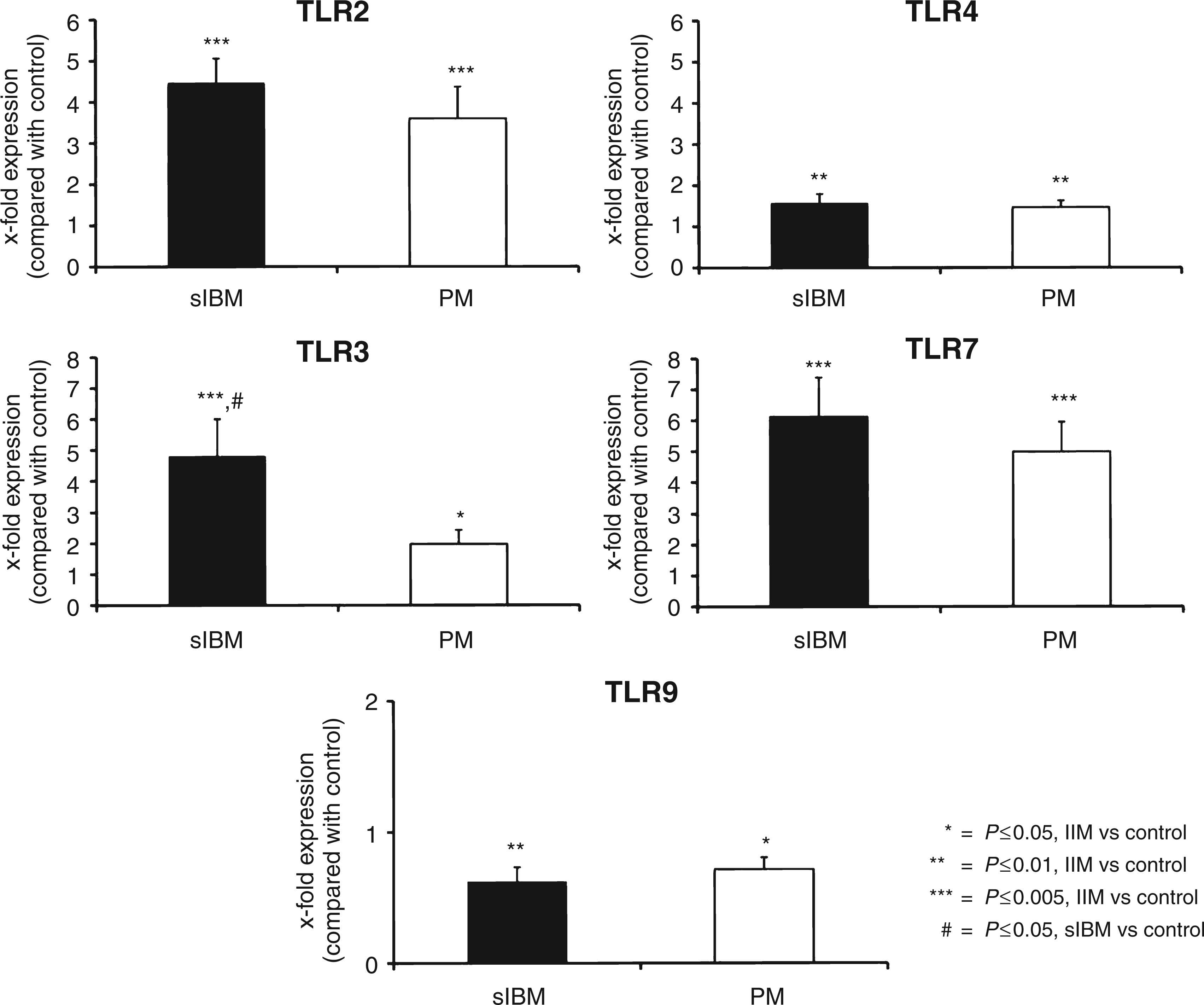
To further analyse the expression pattern of TLR3 in sIBM and PM, and its association with HBD-3, co-localization studies with TLR3 and HBD-3 were performed. Cryosections of three patients with sIBM and PM each and two healthy controls were double-labelled with the aforementioned Abs against HBD-3 and TLR3. Quantification analysis showed a mean value of co-localization (yellow signal) of 66.53% in sIBM and 86.29% in PM (P = 0.006). Expression of HBD-3 without TLR3 (green signal) showed a mean value of 1.41% in sIBM and 5.19% in PM (P = 0.06), and expression of TLR3 without HBD-3 (red signal) a mean value of 22.32% in sIBM and 2.64% in PM (P = 3.5 × 10−5). These data showed that in sIBM, TLR3 was significantly less stronger associated with HBD-3 than in PM (Figure 7). TLR3 was expressed in cellular infiltrations and necrotic muscle fibres in both IIM. Moreover, TLR3 could be detected forming ‘aggregate-like’ structures in the centre of muscle fibres in sIBM, which co-localized to HBD-3-expression (Figure 7).
Co-localization of HBD-3 with TLR3. Fresh-frozen sections of sIBM, PM and normal control muscle biopsies (each n = 3 patients with each 10 HPF) were double-labelled with HBD-3 (green) and TLR3 (red), and analysed by confocal laser scanning microscopy. Nuclei were stained with DAPI (blue). Co-localization of HBD-3 and TLR3 is shown in pixels (%/HPF) in sIBM (black bar) and PM (white bar). Pixels exclusively lightened green or red were shown as HBD-3 + or TLR3 + respectively. Scale bar = 100 µm.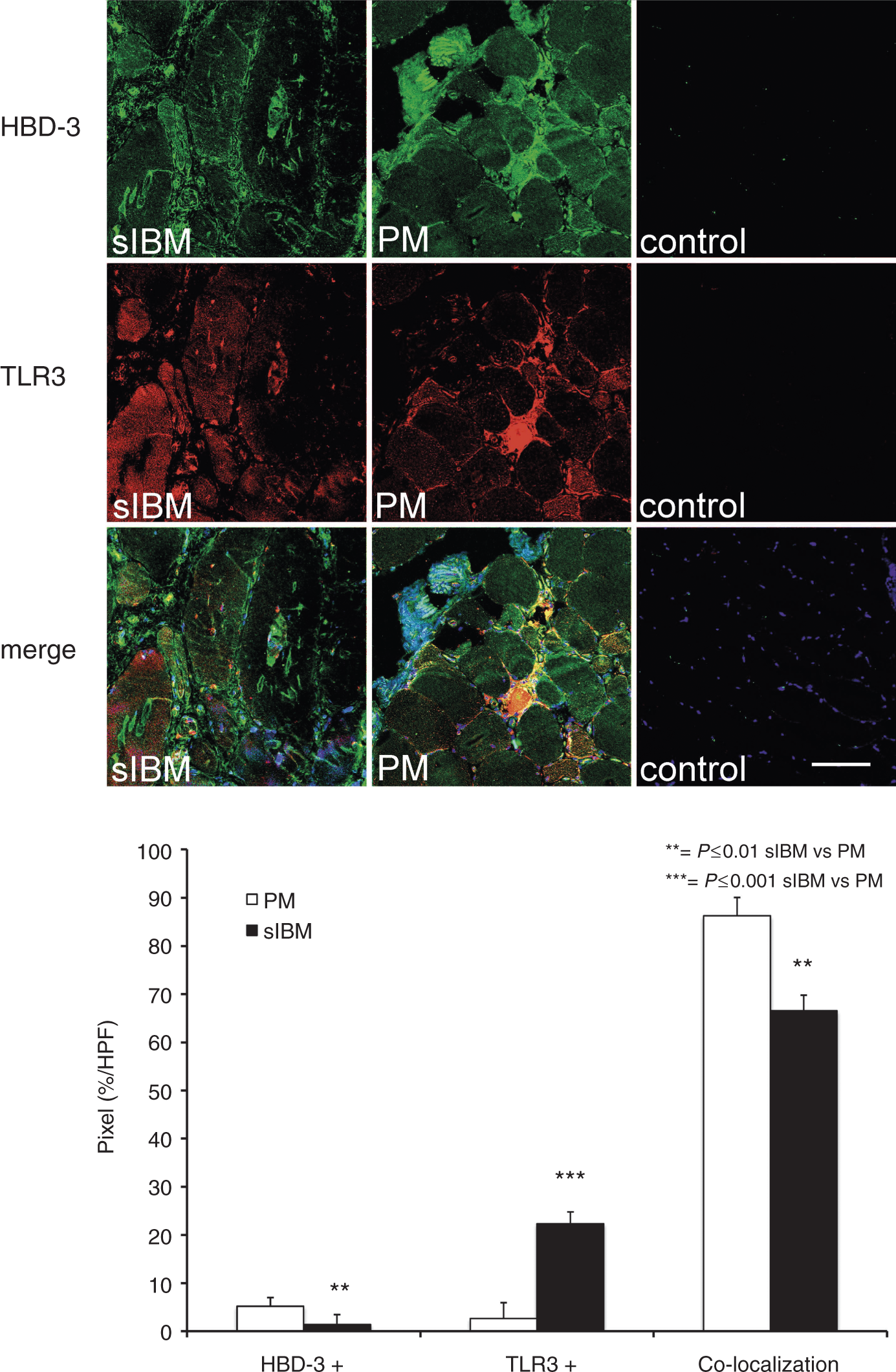
Discussion
In this study we investigated the potential role of HDPs as markers of the innate immune system in the pathogenesis of sIBM and PM. We found that, in contrast to other HDPs, expression levels of HBD-3-specific mRNA were increased in sIBM and PM. Immunofluorescence studies depicted a specific distribution pattern of HBD-3 in sIBM. HBD-3 accumulated inside muscle fibres in an ‘aggregate-like’ manner. These central accumulations could only be found in sIBM muscle tissue. In these accumulations, HBD-3 co-localized to intracellular accumulations of LC3 and β-amyloid in sIBM. Additionally, extracellular expression of HBD-3 in inflammatory infiltrations indicated by perimysial CD8+ T-cells, as well as near macrophages invading necrotic fibres could be observed in both IIM. Expression levels of TLR2, TLR4, TLR7 and TLR9 were increased in both IIM, but TLR3 was significantly overexpressed in sIBM compared with PM. Moreover, TLR3 could be displayed in ‘aggregate-like’ structures in the centre of muscle fibres in sIBM, which co-localized to HBD-3.
HBD-3 was associated with active inflammation and showed a characteristic intracellular ‘aggregate-like’ accumulation in sIBM
Our data showed that, among the HDPs, only HBD-3 was overexpressed in sIBM and PM. Furthermore, it was up-regulated in inflammatory infiltrates in both myositis subtypes. Moreover, HBD-3 co-localized to CD8-positive T-cells in both myopathies, although, in PM, the association of CD8-positive T-cells and HBD-3 was significantly stronger.
The association of HBD-3 with inflammatory cells has been studied before. HBD-3 induces chemotaxis in T-lymphocytes and macrophages via interaction with CCR2 and CCR6.11,14,15 Therefore, it seemed likely that a co-localization of HBD-3 with CD8+ T-cells and macrophages close to inflammatory infiltrates contributed, at least in part, to the induction of chemotaxis.
Previous studies also showed different expression patterns of inflammatory markers like CXCL-9, IFN-γ or IL-1β in sIBM and PM. In sIBM, expression levels of these markers were increased within muscle fibres, whereas in PM expression was detected in connective tissue.5,7,22 Moreover, close interrelations between inflammatory and degenerative, as well as autophagic processes in sIBM have been described.7,10,23 In a human muscle cell line, endogenous APP was targeted to LC3-positive autophagosomes. In muscle fibres of sIBM patients, an increase in autophagic activity, often associated with an increase in inflammatory activity, could be demonstrated. 10 It has also been shown that TNF-α and IFN-γ lead to increased macro-autophagy in myofibres and to an increase of antigen presentation by MHC II molecules. These alterations were strongly associated with cellular infiltrations of CD4+ and CD8+ T-cells. 23 Furthermore, it is known that host defence peptides are associated with endo- and autolysosomal processes.24,25 The co-localization of HBD-3 with β-amyloid and LC3 therefore indicated a link of inflammatory HBD-3 to degenerative alterations and autophagic processes. Thus, associated with the characteristic pathological alterations in sIBM, HBD-3 may possibly serve as a suitable marker and diagnostic tool in sIBM. However, its specificity and sensitivity have not been analysed, and, owing to the low number of samples and lack of controls, the role of HBD-3 as a possible diagnostic parameter remains to be investigated in larger studies.
Overexpression of endosomal TLR3 in sIBM contributed to its pathophysiologic characteristics
As HBD-3 is induced by TLR-dependent pathways, expression levels of a specific subset of TLRs involved in inflammatory myopathies were analysed. An overexpression of the same subset of TLRs (2, 3, 4 and 7), as well as a decrease of TLR9 expression was measured in both IIM, showing a general increase of inflammatory activity. Increased expression levels of TLR2, TLR4 and TLR9, 19 as well as TLR7, and also for TLR3, 20 have already been described in PM and DM. A further study by Brunn et al. 26 showed slightly increased expression levels for TLR9 in sIBM and PM patients compared with controls. At present we have no explanation for these differences between the studies. Owing to the smaller group sizes in the other two studies, a clear assessment is also difficult.
Considering overexpression of TLR3, we measured different expression levels, which showed a significant higher expression level in sIBM compared with PM. The intracellular localization pattern of TLR3 in sIBM resembled the intracellular accumulation of LC3 in sIBM. This result was of special interest considering the particular intracellular localization and activation of TLR3. TLR3 is located in the endosome, and induced by host or viral dsRNA and cell debris. It is assumed to play a key role in antiviral defence and leads to increased autophagic activity.16,27 Moreover, overexpression of TLR3 is associated with an increased uptake of β-amyloid (Aβ42) in microglia, 28 and previous studies described overexpression of TLR3 in sIBM.29,30 Moreover, it has been shown that TLR activation contributes to induction of p62, a key factor of autophagy. 31 Considering sIBM-specific histopathological changes, i.e. accumulation of β-amyloid and increased autophagic activity7,9,10,23 it is interesting that endosomal TLR3 was overexpressed in sIBM muscle tissue, but showed a significantly lower amount of co-localization to HBD-3. Activation of TLR3 in sIBM might indicate increased endosomal autophagy in sIBM. The exact role and interactions of HBD-3, β-amyloid, LC3 and TLR3 in sIBM pathophysiology, however, is currently unknown and needs further investigation.
The close relationships between inflammatory and degenerative, as well as autophagic processes in sIBM once again raised the question of the causative pathological aberration. Although the complex interactions between degenerative and inflammatory alterations in sIBM have been studied widely before, the initial trigger of the disease is still not known. In our opinion, activation of the innate immune system is not the primary pathological process in sIBM, but rather a secondary effect due to degradation processes activating inflammatory reactions in sIBM. In this context, the effect of host RNA on TLR3-expression and increased autophagic and inflammatory activity may be of importance in sIBM pathogenesis. However, the dynamic interaction of the different metabolic pathways concerning autophagy, the role of TLR-dependent pathways and the activation of the innate immune system will have to be investigated in further studies.
Footnotes
Acknowledgements
We sincerely thank A. Schreiner for excellent technical support.
Funding
This work was supported by the medical faculty of the Ruhr University Bochum (FoRUM; F700-2010).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.