
Abstract
NLRX1 is a member of the Nod-like receptor family of intracellular sensors of microbial- and danger-associated molecular patterns. NLRX1 has a N-terminal mitochondrial addressing sequence that localizes the protein to the mitochondrial matrix. Recently, conflicting reports have been presented with regard to the putative implication of NLRX1 as a negative regulator of MAVS-dependent cytosolic antiviral responses. Here, we generated a new NLRX1 knockout mouse strain and observed that bone marrow-derived macrophages and murine embryonic fibroblasts from NLRX1-deficient mice displayed normal antiviral and inflammatory responses following Sendai virus infection. Importantly, wild type and NLRX1-deficient mice exhibited unaltered antiviral and inflammatory gene expression following intranasal challenge with influenza A virus or i.p. injection of Poly (I:C). Together, our results demonstrate that NLRX1 does not participate in the negative regulation of MAVS-dependent antiviral responses.
Introduction
Nod-like receptors (NLRs) are a family of cytosolic pattern recognition molecules (PRMs), which trigger the activation of innate immune pathways following the detection of conserved microbial- and danger-associated molecular patterns.1–3 In humans, NLRs represent one of the major groups of intracellular PRMs, but the specific function of several NLR proteins in innate immunity remains poorly characterized. NLRX1 is a unique NLR in that it displays a N-terminal mitochondrial addressing sequence.4,5 Overexpression of NLRX1 has been shown to amplify inflammatory pathways through the induction of reactive oxygen species (ROS).5,6 In resting conditions, endogenous NLRX1 localizes to the mitochondrial matrix, where the protein is matured by matrix protein peptidases and interacts with UQCRC2, a matrix-facing protein of the respiratory chain complex III.7,8 In addition, NLRX1 was also identified as a negative regulator of antiviral signalling induced by the RIG-I like receptor (RLR) pathway through a direct interaction with the cytosol-facing mitochondrial adaptor protein MAVS (also known as Cardif, IPS-1 or VISA), 4 a critical adaptor protein of RLR signalling that is associated with the mitochondrial outer membrane.9–12
Recently, NLRX1-deficient mice were generated by two independent groups to explore the role of NLRX1 in host responses to viruses and TLR ligands, such as LPS.8,13 Allen et al. reported that NLRX1-deficient mice and NLRX1-deficient mouse embryonic fibroblasts (MEFs) displayed increased activation of type I interferon responses following viral infection compared with their wild type (WT) counterparts. 13 This effect of NLRX1 appeared to be cell-type specific, as RIG-I-dependent antiviral signalling was similar in NLRX1-deficient and WT bone marrow derived macrophages (BMDMs). 13 In contrast, Rebsamen et al. reported that NLRX1-deficient MEFs and BMDMs had normal antiviral cytokine production in response to virus infection or double-stranded (ds) RNA stimulation, and also observed that induction of IFN-β and IL-6 in response to i.v. injection of dsRNA was not affected by NLRX1 deficiency. 8 Finally, a third group generated NLRX1-knockdown mice and proposed that NLRX1 could modulate antiviral responses to vesticular stomatitis virus in vitro, but not in vivo. 14
In addition to the effect on MAVS-dependent antiviral responses, Allen et al. and Xia et al. also proposed a more general role for NLRX1 as a negative regulator of TRAF6-dependent NF-κB signalling.13,14 Accordingly, NLRX1-deficient cells and mice were found to display exacerbated responses to LPS, which is known to signal to NF-κB via TLR4 through TRAF6.15,16 Mechanistically, LPS stimulation was shown to trigger NLRX1 ubiquitination and dissociation from TRAF6, while ubiquitinated NLRX1 would stably interact with the IκB kinase (IKK) complex to inhibit NF-κB activation. 14 How the mitochondrial matrix protein NLRX1 could physically interfere with the TRAF6/IKK complex in the cytosol was not investigated in these studies.
As presented above, conflicting results have been reported with regard to the role of NLRX1 in MAVS-dependent antiviral signalling in two independently generated strains of NLRX1-deficient mice. Therefore, we aimed to re-assess this proposed role by ex vivo and in vivo analysis of a third independent strain of NLRX1-deficient mice. No differences were observed in the levels of antiviral (IFN-α, IFN-β, STAT-2, IRF-7 and CXCL10) and inflammatory (IL-6 and KC) gene expression between WT or NLRX1-deficient cells (MEFs and BMDMs) infected with Sendai virus. Moreover, phosphorylation of IRF-3, a key event in MAVS-dependent antiviral signalling, was similar in WT and NLRX1-deficient MEFs infected with Sendai virus, and secretion of IL-6, CXCL10, KC and IFN-β in response to Sendai virus infection was unaltered in NLRX1-deficient MEFs compared with WT MEFs. Finally, in vivo studies confirmed that the early inflammatory and antiviral signalling was normal in NLRX1-deficient mice, following intranasal challenge with influenza A virus or i.p. administration of the viral dsRNA mimetic Poly (I:C). Together, these results demonstrate clearly that NLRX1 does not act as a negative regulator of MAVS-dependent antiviral signalling.
Materials and methods
Cell culture and reagents
MEF cells were cultured in DMEM (Wisent, St-Bruno, Quebec, Canada) supplemented with 10% heat-inactivated FBS (Wisent) and 1% penicillin/streptomycin. Cells were maintained in 95% air and 5% CO2 at 37℃.
MEF and BMDM preparations
MEFs and BMDMs from WT and NLRX1-deficient mice were isolated in accordance with protocols approved by the University of Toronto using standard protocols. All experiments involving MEFs were performed using low passage primary cells (fewer than four passages). Freshly isolated BMDMs were prepared as described previously
17
and seeded in 10-cm dishes with complete culture medium [RPMI supplemented with 2 mM
RNA isolation and real-time quantitative PCR
Total RNA was extracted using a GeneJET™ RNA Purification Kit (Fermentas, Burlington, Ontario, Canada) and treated with Turbo DNase (Ambion, Austin, TX, USA) to remove traces of genomic DNA. One microgram of purified RNA was then reverse transcribed using M-MLV Reverse Transcriptase (Sigma, Oakville, Ontario, Canada) with random hexamer and oligo-dT primers (Fermentas). cDNA was diluted accordingly and 20 µl reactions were set up using Power SYBR Green Master Mix 2X to run quantitative PCR (qPCR). Raw cycle threshold (CT) values were obtained from an ABI 7900HT qPCR machine. Results were analysed using the 2 −ΔCt formula normalizing target gene expression to mRPL-19. qPCR primer sequences are presented in the following list. Lung tissue RNA was extracted using a RNA Tissue Miniprep Kit (Geneaid, Shijr City, New Taipei, Taiwan).
The real-time qPCR primers were designed using Universal ProbeLibrary (Roche, Laval, Quebec, Canada) and the sequences were the following:
mRPL-19-For: GCATCCTCATGGAGCACAT mRPL-19-Rev: CTGGTCAGC CAGGAGCTT mNLRX1-For:TGCCATTTGCCCAGGACCTC TT mNLRX1-Rev: GCTCCACTGGATCAAGAAG GAGATATGC mIFN-β-For: TGAACTCCACCAGCAGACAG mIFN-β-Rev: ACCACCATCCAGGCGTAGC mIFN-α-For: TCAAGCCATCCTTGTGCTAA mIFN-α-Rev: GTCTTTTGATGTGAAGAGGTT CAA mSTAT-2-For: GGAACAGCTGGAACAGTG G T mSTAT-2-Rev: GTAGCTGCCGAAGGTGGA mIRF-7-For: CTGGAGCCATGGGTATGCA mIRF-7-Rev: AAGCACAAGCCGAGACTGCT mCXCL10-For: ATCATCCCTGCGAGCCTATC CT mCXCL10-Rev: GACCTTTTTTGGCTAAACG CTTTC mRANTES-For: GCCCACGTCAAGGAGTATT TCTA mRANTES-Rev: ACACACTTGGCGGTTCCT T C mIL-6-For: CCAGAAACCGCTATGAAGTTCC mIL-6-Rev:TTGTCACCAGCATCAGTCCC KC-For: ACTGCACCCAAACCGAAGTC KC-Rev: CAAGGGAGCTTCA GGGTCAA mTNF-α-For: TCCCAGGTTCTCTTCAAGGGA mTNF-α-Rev: GGTGAGGAGCACGTAGTCGG mATF3-For: CGAAGACTGGAGCAAAATGA TG mATF3-Rev: CAGGTTAGCAAAATCCTCAAA TAC mIFN-γ- For: ATCTGGAGGAACTGGCAAAA mIFN-γ- Rev: TTCAAGACTTCAAAGAGTCTG AGGTA Influenza A-For: GGCCCAACCACAACACAA AC Influenza A-Rev: AGCCCTCCTTCTCCGTCAGC.
Western blot and Abs
Total cell lysates were prepared and run on SDS-PAGE using standard techniques. Gels were transferred onto polyvinylidene fluoride membrane using a semi-dry transfer apparatus and blocked in 5% skim milk containing TBS/T (0.1% Tween 20). Primary and secondary Abs were diluted in TBS/T and incubated for 1 h. The following Abs were used to for protein detection: mouse monoclonal anti-tubulin (Sigma, T9026; 1:10,000), rabbit monoclonal phospho-IRF-3 (Ser396, 4D4G; Cell Signalling, #4947; 1:1000) and rabbit polyclonal anti-NLRX1 (Proteintech, 17 215-1-AP; 1:1,000) to detect endogenous murine NLRX1.
Generation of NLRX1-deficient mice
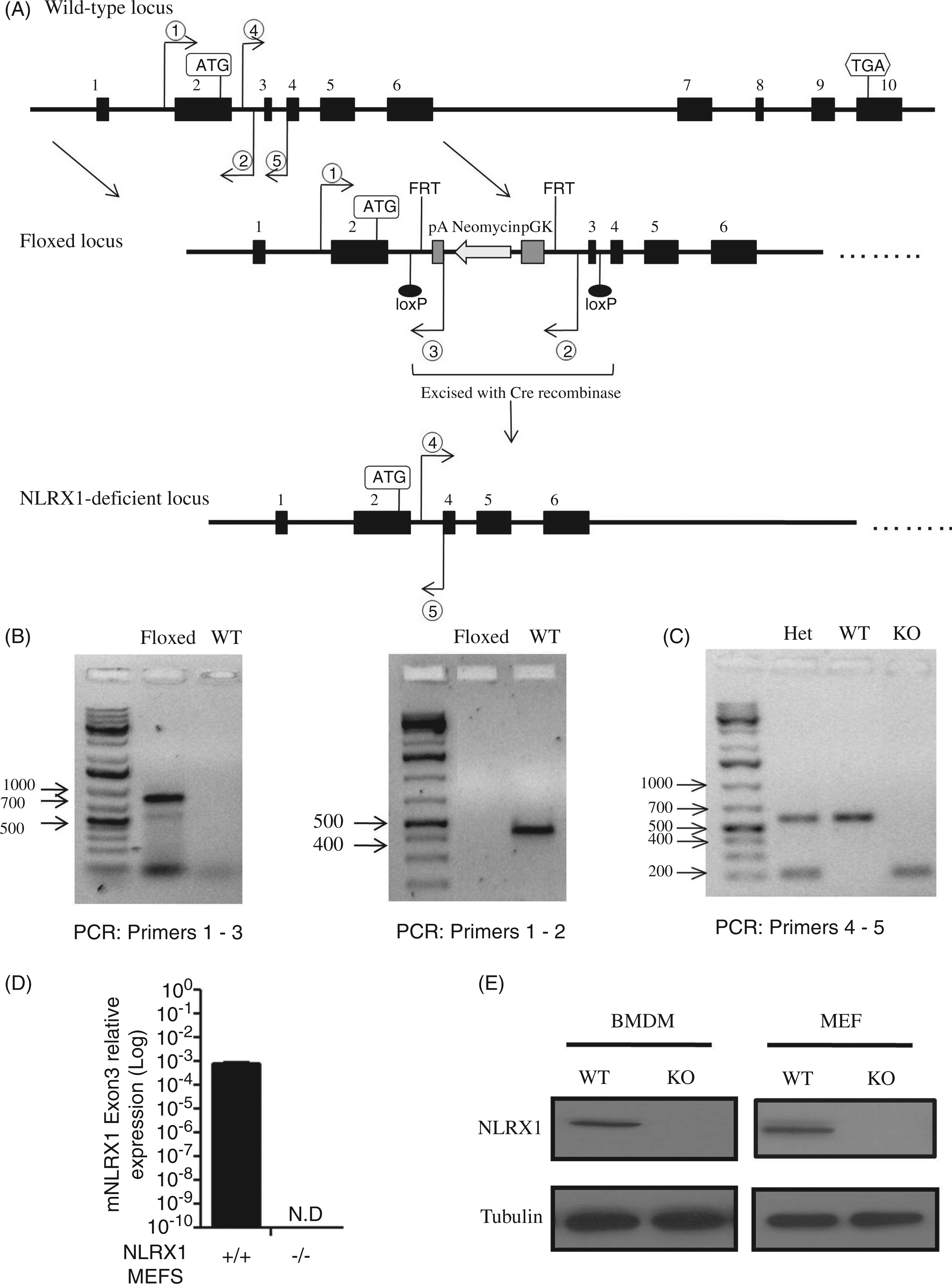
A scheme detailing the generation of C57Bl/6 NLRX1-deficient mice is shown in Figure 1A. Floxed mice were generated by inserting a phosphoglycerate kinase-neomycin (PGK-neo) selection cassette upstream of Exon 3 (Ozgene, Bentley DC, WA, Australia) resulting in the generation of loxP sites flanking Exon 3. Floxed mice were then crossed with a Cre mouse [C57Bl/6 mouse expressing Cre recombinase under a constitutive promoter from Jackson Laboratories (Bar Harbor, ME, USA)] to generate the NLRX1-deficient mouse lacking Exon 3. The following PCR primers (5’-3’) were used to genotype floxed and NLRX1-deficient mice:
Primer 1—(CCA TTT GCC AAT CCC ACT CAC) Primer 2— (ACC AAG AAC CTA ACC CAC GGT C) Primer 3— (TTG CCA GCC ATC TGT TGT TTG) Primer 4— (GGC CCT CTT TTA GCT GTT TCA) Primer 5— (CGA TCC CAT TCC CCA TTA TT).
Generation of NLRX1-deficient mice. (A) Locus map of mouse NLRX1 gene illustrating the 10 exons found within the transcript (NM_178420.2). Floxed mice were screened for the presence of a PGK-Neo selection cassette inserted upstream of exon 3 that is flanked by a flippase recombinase site (FRT). Exon 3 is flanked by loxP sites in the floxed mice and was excised when crossed with a Cre mouse (C57Bl/6 mice expressing Cre recombinase under a constitutive promoter) to generate the NLRX1-deficient mouse. (B) Floxed mice containing the PGK-Neo selection cassette were screened by PCR using two different primer sets (1–3 and 1–2). (C) Absence of Exon 3 in NLRX1-deficient mice was genotyped by PCR using primers 4–5. (D) qPCR on cDNA prepared from WT and NLRX1-deficient MEFs. A primer specific for exon 3 of mNLRX1 was designed to measure mNLRX1 expression. N.D., not detectable. (E) Lysates from WT and NLRX1-deficient MEFs and BMDMs were analysed by Western blot using an anti-NLRX1 Ab that detects the endogenous murine protein.
All animal procedures were conducted in compliance with regulations from the University of Toronto.
Viruses and in vitro infections
The Sendai H4 virus strain (SeV-H4) has been described previously.18,19 Encephalomyocarditis virus (EMCV) strain K-2 was from Y. Svitkin (McGill, Montreal) and vesicular stomatitis virus (VSV) expressing GFP was from J. Perrault (San Diego State University). MEFs and BMDMs were seeded in 6-well tissue culture plates the day before and incubated with viruses for 1 h in serum-free medium. Cells were then rinsed with 1 × PBS, and fresh medium containing FBS was added for the duration of the infection.
In vivo influenza A infection and Poly (I:C) administration
WT and NLRX1-deficient mice were anaesthetized and challenged intranasally (30 µl volume) with the Puerto Rico/8/34 (PR8) strain of influenza A virus. Mice were sacrificed 72 h post-infection and whole lungs were harvested for total RNA extraction. Bronchoalveolar lavage was collected by washing the trachea with PBS. Poly (I:C) was purchased from Invivogen (San Diego, CA, USA) and 200 µg/mouse was injected i.p. in a volume of 200 µl. Spleens were harvested 4 h post-injection and total RNA was prepared.
Cytokine and IFN-β determination
KC, mIL-6 and mCXCL10 amounts in the supernatant of WT and NLRX1-KO MEFS were quantified by ELISA (R&D Systems, Minneapolis, MN, USA) following the manufacturer’s instructions. Similarly, KC, mCXCL10 and mRANTES amounts in bronchoalveolar lavage of WT and NLRX1-KO mice were measured by ELISA (R&D Systems). B16-Blue™ IFN-α/β cells (kindly provided by Dr Maziar Divangahi, McGill University, Montreal) were used to detect mIFN-β following the manufacturer’s instructions. In brief, supernatants from WT and NLRX1-KO MEFS were added to a 96-well plate seeded with 75,000 B16-Blue™ IFN-α/β cells containing the secreted alkaline phosphatase (SEAP) reporter gene under the control of an IFN-α/β ISG54 promoter. Cells were incubated for 20 h at 37℃ in a CO2 incubator and 20 µl of supernatant was removed to measure SEAP activity. p-Nitrophenyl phosphate substrate (1 mg/ml; BioShop Canada Inc., Burlington, Ontario, Canada) was prepared fresh in development buffer (100 mM Tris-HCl, pH 9.5, 100 mM NaCl, 5 mM MgCl2) and added to each sample. The mixture was incubated at 37℃ for 10 min or until a yellow colour appeared. The absorbance was measured at 405 nm using the Victor3 1420 multilabel counter (Perkin Elmer, Waltham, MA, USA).
Statistical analysis
Prism (GraphPad, San Diego, CA, USA) software was used to plot data and a Student’s t-test was applied to determine statistical significance. A P-value of < 0.05 was considered to be statistically significant.
Results
NLRX1-deficient cells exhibit normal antiviral responses
In order to clarify the potential role of NLRX1 in antiviral immunity, we generated a new strain of NLRX1-deficient mice on a C57Bl/6 background (Figure 1A). NLRX1-floxed mice were generated and screened by PCR for the presence of a PGK-neomycin cassette inserted upstream of Exon 3 (Figure 1B). NLRX1-floxed mice, which contained Exon 3 flanked by loxP sites, were then crossed with C57Bl/6 mice expressing Cre recombinase under a constitutive promoter, and deletion of Exon 3 was screened by PCR to identify NLRX1-deficient mice (Figure 1C). qPCR was performed using a mouse NLRX1 Exon 3 specific primer to confirm the absence of mNLRX1 mRNA expression in NLRX1-deficient MEFs (Figure 1C). Likewise, NLRX1 protein expression was abolished in NLRX1-deficient MEFs and BMDMs as measured by Western blotting using an NLRX1 Ab that detects the endogenous murine NLRX1 (Figure 1D).
Next, we assessed antiviral gene expression in WT and NLRX1-deficient BMDMs (Figure 2A–H) infected with Sendai virus for 3 h or 6 h, and qPCR was performed to measure the expression of a panel of the antiviral and inflammatory genes mIFN-β (Figure 2A), mIFN-α (Figure 2B), mSTAT-2 (Figure 2C), mIRF-7 (Figure 2D), mCXCL10 (Figure 2E), mRANTES (Figure 2F), mIL-6 (Figure 2G) and KC (Figure 2H). In none of the conditions and genes tested did we identify a statistically significant difference in host response to Sendai virus between WT and NLRX1-deficient cells.
Antiviral gene expression is unaffected in NLRX1-deficient BMDMs. (A–H) WT and NLRX1-deficient (NLRX1-KO) BMDMs were infected with Sendai virus for the indicated time points and total RNA was extracted. cDNA was prepared and qPCR was performed to evaluate expression of mIFN-β (A), mIFN-α (B), mSTAT-2 (C), mIRF-7 (D), mCXCL10 (E), mRANTES (F), mIL-6 (G) and KC (H). Each data point plotted on graphs represents the result of an independent experiment for that condition. At least three independent experiments are plotted together for each target gene and condition assayed. No statistical differences were observed among the evaluated genes (two-tailed unpaired t-test, P < 0.05).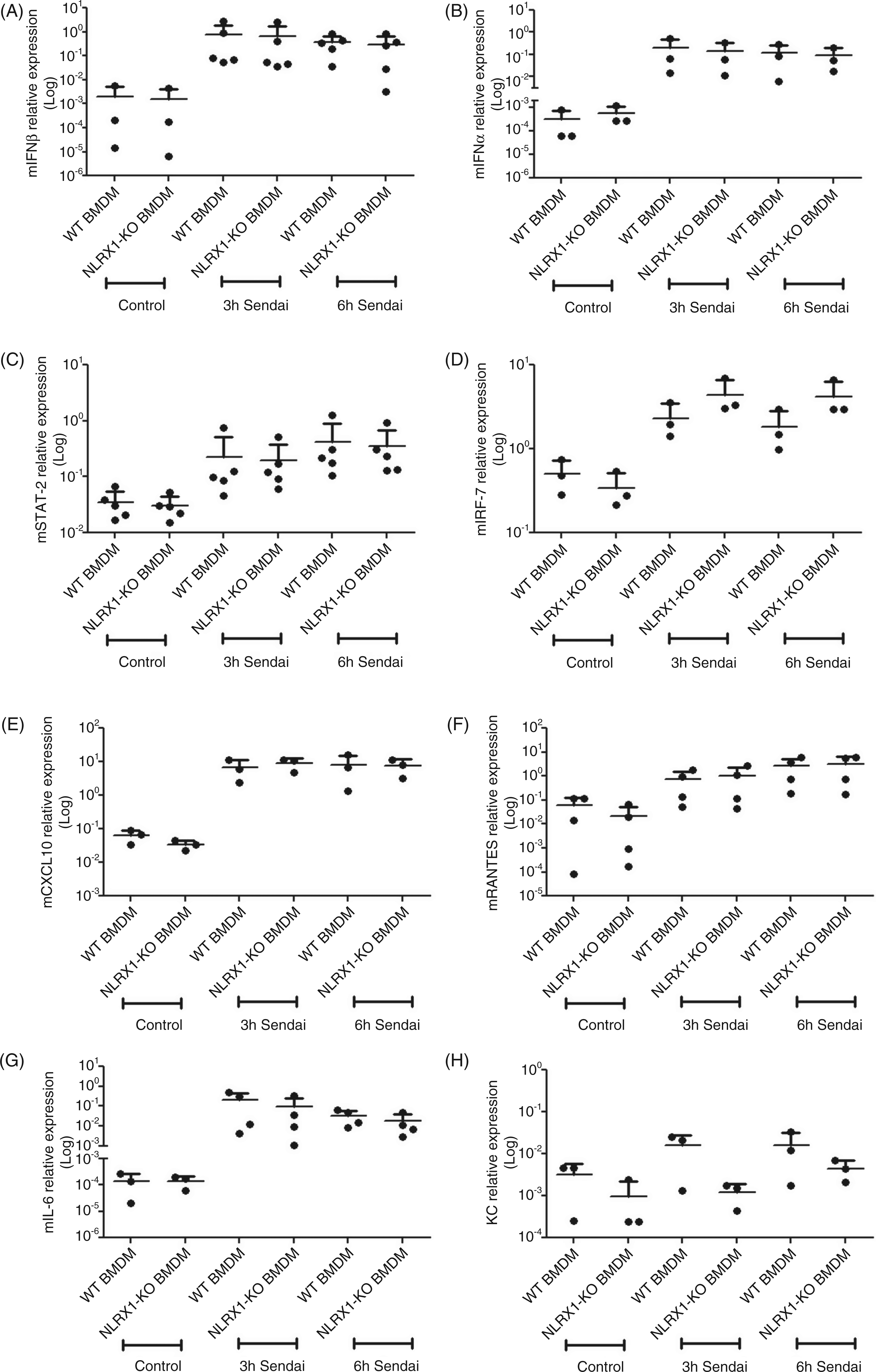
We then followed antiviral responses to Sendai virus infection in WT and NLRX1-deficient MEFs (Figure 3A–L). Using qPCR, we first showed that infection with Sendai virus for 3 h or 6 h resulted in comparable induction of mIFN-β (Figure 3A), mIFN-α (Figure 3B), mSTAT-2 (Figure 3C), mCXCL10 (Figure 3D), mIRF-7 (Figure 3E), mIL-6 (Figure 3F) and KC (Figure 3G) in WT and NLRX1 knockout cells. Using ELISA we further observed similar secretion of mIL-6 (Figure 3H), mCXCL10 (Figure 3I) and KC (Figure 3J) in WT and NLRX1 knockout MEFs infected with Sendai virus for 1–4 h. Relative levels of IFN-β secretion were also determined using a reporter system in B16-Blue™ IFN-α/β cells, and, again, similar levels of IFN-β secretion were noted between WT and NLRX1-deficient cells infected for 1–4 h with Sendai virus (Figure 3K). Finally, phosphorylation of IRF-3, a key event in MAVS-dependent induction of IFN-α/β in virus-infected cells, was determined by Western blotting in lysates of WT and NLRX1-deficient MEFs infected for 1–4 h with Sendai virus, and similar induction of IRF-3 phosphorylation was observed in lysates of infected WT and NLRX1 knockout MEFs (Figure 3L).
Loss of NLRX1 in MEFs does not affect antiviral responses during Sendai virus infection. Wild type (WT) and NLRX1-deficient (NLRX1-KO) MEFs were infected with Sendai virus and total RNA was extracted at 3 h and 6 h. cDNA was prepared and antiviral gene expression was evaluated by qPCR for mIFN-β (A), mIFN-α (B), mSTAT-2 (C), mCXCL10 (D), mIRF-7 (E), IL-6 (F) and KC (G). Each data point was obtained from an independent experiment for that condition. At least 3 (and up to 10 for certain genes) independent experiments are plotted together for each target gene and condition assayed. No statistical differences were observed among the evaluated genes (two-tailed unpaired t-test, P < 0.05). (H–J) Supernatants from WT and NLRX1-KO MEFS infected with Sendai (1–4 h) were collected and cytokine production was measured by ELISA for mIL-6 (H) mCXCL10 (I) and KC (J). The results presented are from one experiment representative of three independent experiments. (K) Supernatants from (H–J) were added to B16-Blue™ IFN-α/β cells and mIFN-β was determined by measuring secreted alkaline phosphatase (SEAP), induced by the IFN-α/β ISG54 promoter. SEAP activity was measured at 405 nm when the substrate was provided. The results presented are from one experiment representative of three independent experiments. No statistical differences were observed among the evaluated cytokines (two-tailed unpaired t-test, P < 0.05). (L) WT and NLRX1-KO MEFS were infected with Sendai virus (1 h, 2 h and 4 h) and cell extracts were prepared to examine phospho-IRF-3 expression by Western blotting. NLRX1 expression was also assessed and tubulin was used as a loading control. Results from one experiment representative of four independent experiments is shown. CTR, control; OD, optical density.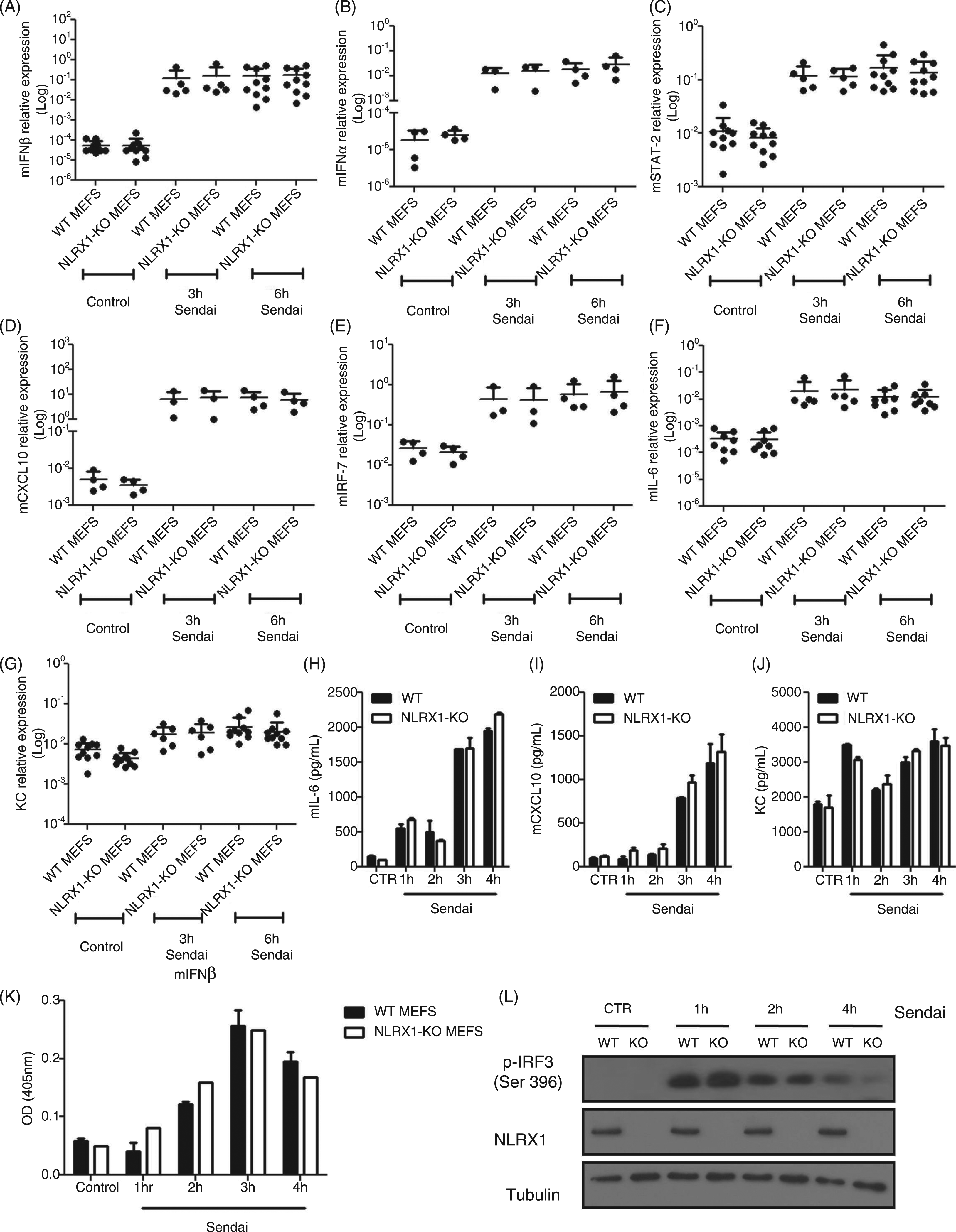
In order to determine if normal antiviral responses to other viruses occurred in NLRX1-deficient cells, WT and NLRX1-deficient MEFs were infected for 3 h or 6 h with EMCV or VSV, two RNA viruses that trigger MAVS-dependent antiviral responses in infected cells. Again, comparable induction of mIFN-β was observed in WT and NLRX1-deficient MEFs infected for 3 h or 6 h with VSV (Figure 4A) or EMCV (Figure 4B).
IFN-β gene expression remains unaltered in NLRX1-deficient MEFs following VSV and EMCV infection. WT and NLRX1-deficient MEFs were infected with VSV-GFP (A) or EMCV (B), and IFN-β gene expression was measured by qPCR. Each data point was obtained from an independent experiment and at least three independent experiments are plotted. No statistical differences were observed (two-tailed unpaired t-test, P < 0.05).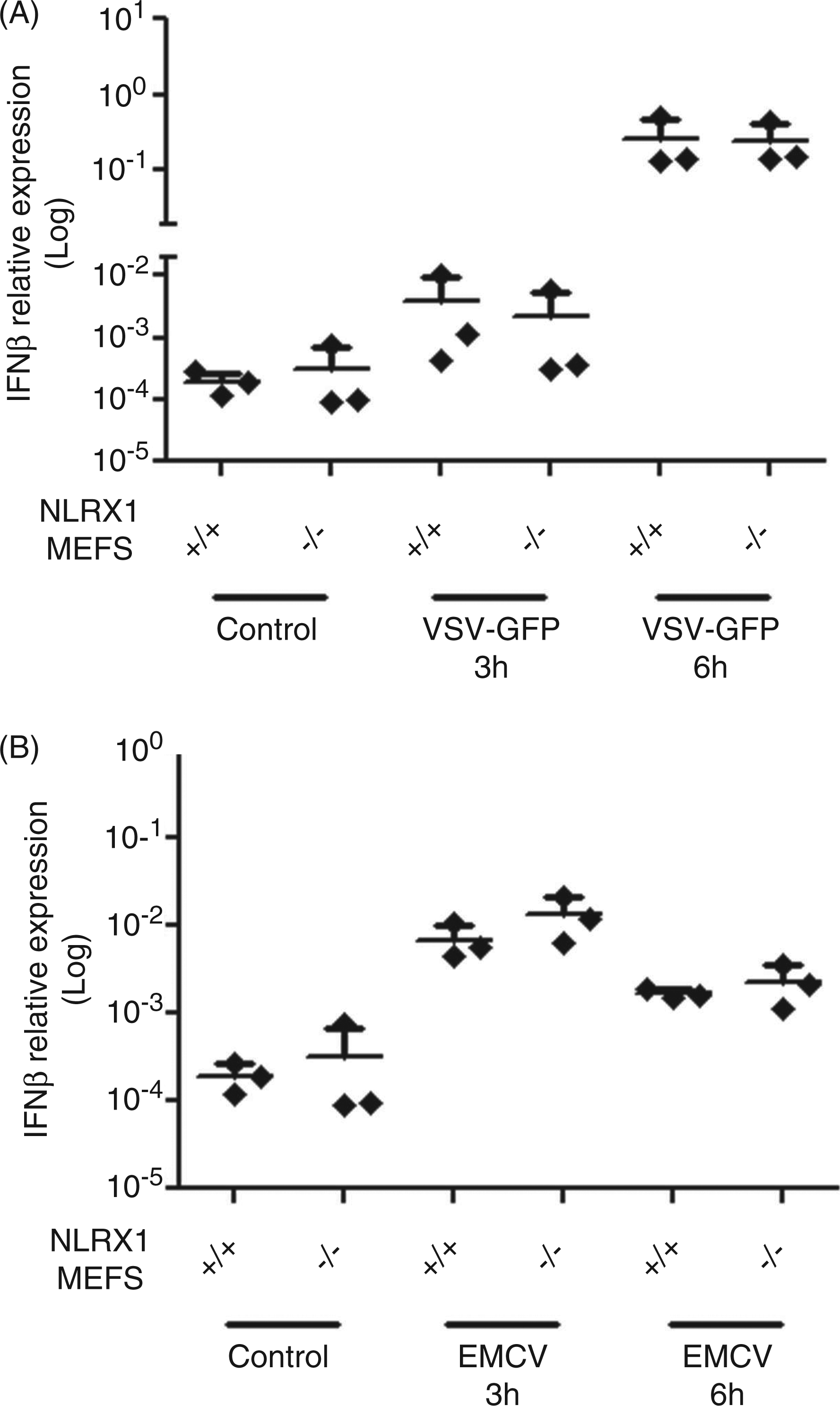
Together, these observations demonstrate that MAVS-dependent induction of host antiviral responses is normal in NLRX1-deficient cells.
NLRX1-deficient mice have normal antiviral responses to Poly (I:C) and influenza virus infection
The PR8 strain of influenza A virus is a respiratory pathogen that is capable of inducing a robust antiviral response in mice. We aimed to investigate antiviral and inflammatory gene and protein expression in NLRX1-deficient mice infected with influenza virus in vivo. To do so, WT and NLRX1-deficient mice were intranasally infected with influenza virus PR8 and sacrificed 72 h post-infection. Lung tissue was then extracted and qPCR performed to measure host gene expression in response to viral challenge. Strong host response to viral challenge was observed, as evidenced by the substantial induction of mIFN-β (Figure 5A), mIFN-α (Figure 5B), mIFN-γ (Figure 5C), mCXCL10 (Figure 5D), mIL-6 (Figure 5E), KC (Figure 5F), mRANTES (Figure 5G), mSTAT-2 (Figure 5H), mATF3 (Figure 5I), mTNF-α (Figure 5J) and mIRF-7 (Figure 5K) in the lungs of infected animals (Figure 5A–K). Similar host responses were observed in WT and NLRX1-deficient mice (Figure 5A–K), and comparable levels of infection were obtained in the lungs of WT and NLRX1-deficient mice, as evidenced by the analysis of viral HA gene expression in lung homogenates (Figure 5L). Protein levels of KC (Figure 5M), mCXCL10 (Figure 5N) and mRANTES (Figure 5O) in broncho-alveolar lavages of mice infected with influenza virus PR8 for 72 h were also similar between WT and NLRX1-deficient mice.
In vivo analysis of antiviral responses in NLRX1-deficient mice infected with influenza A. (A–L) WT and NLRX1-deficient mice were challenged intranasally with the PR8 strain of influenza A virus and sacrificed 72 h post-infection. Total RNA was extracted from whole lungs and cDNA was prepared. Antiviral gene expression was evaluated by qPCR for mIFN-β (A), mIFN-α (B), mIFN-γ (C), mCXCL10 (D), mIL-6 (E), KC (F), mRANTES (G), mSTAT-2 (H), mATF3 (I), mTNF-α (J), mIRF-7 (K) and influenza HA gene (L). (M–O) Bronchoalveolar lavage was collected from WT and NLRX1-KO infected mice and cytokine production was measured by ELISA for KC (M), mCXCL10 (N) mRANTES (O). Each data point represents one mouse. Data were pooled from two independent experiments with at least six mice infected for each genotype. No statistical differences were observed among the evaluated genes or cytokines (two-tailed unpaired t-test, P < 0.05). N.D., not detectable.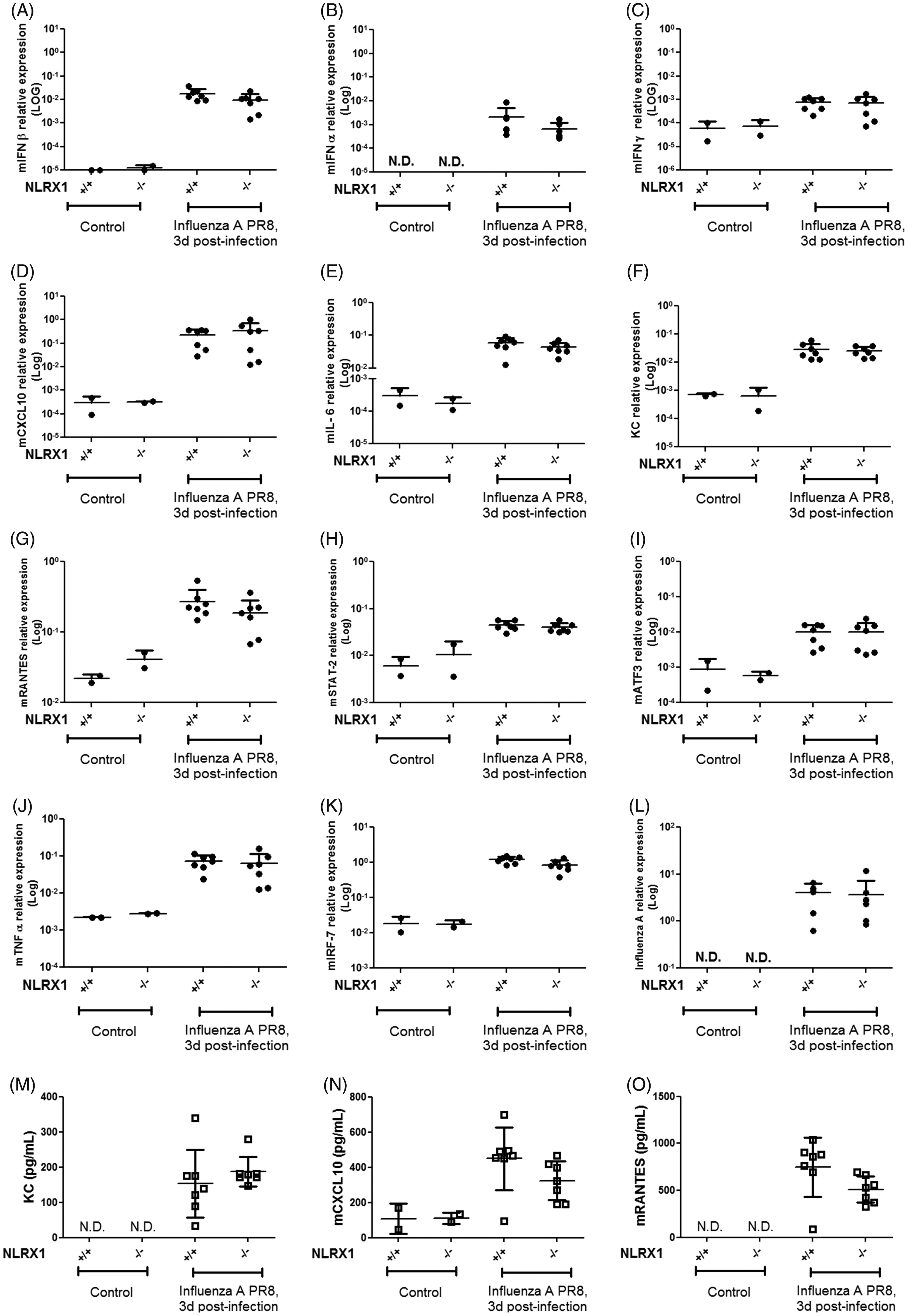
Finally, we investigated the host response of WT and NLRX1 knockout mice to in vivo i.p. injection of Poly (I:C), a synthetic analog of viral dsRNA. To do so we measured antiviral gene expression by qPCR in the spleens of animals 4 h post-i.p. injection of Poly (I:C). Strong antiviral responses were observed, as evidenced by increased expression of mIFN-β (Figure 6A), mIFN-α (Figure 6B), mCXCL10 (Figure 6C), mSTAT-2 (Figure 6D), mIRF-7 (Figure 6E), mRANTES (Figure 6F), mIL6 (Figure 6G), KC (Figure 6H) and mIFN-γ (Figure 6I) in the spleens of Poly(I:C)-injected animals. However, similar inductions were observed in WT and NLRX1-deficient mice (Figure 6A–I).
Poly (I:C)-induced antiviral gene expression is normal in NLRX1-deficient mice. (A–I) WT and NLRX1-deficient mice were stimulated by i.p. injection with Poly (I:C) (200 µg/mouse) for 4 h. Total RNA was extracted from spleen tissue and cDNA was prepared. Expression levels of mIFN-β (A), mIFN-α (B), mCXCL10 (C), mSTAT-2 (D), mIRF-7 (E), mRANTES (F), mIL-6 (G), KC (H) and mIFN-γ (I) were analysed by qPCR. Each data point represents one mouse. Data were pooled from two independent experiments with at least six mice infected for each genotype. No statistical differences were observed among the evaluated genes (two-tailed unpaired t-test, P < 0.05). N.D., not detectable.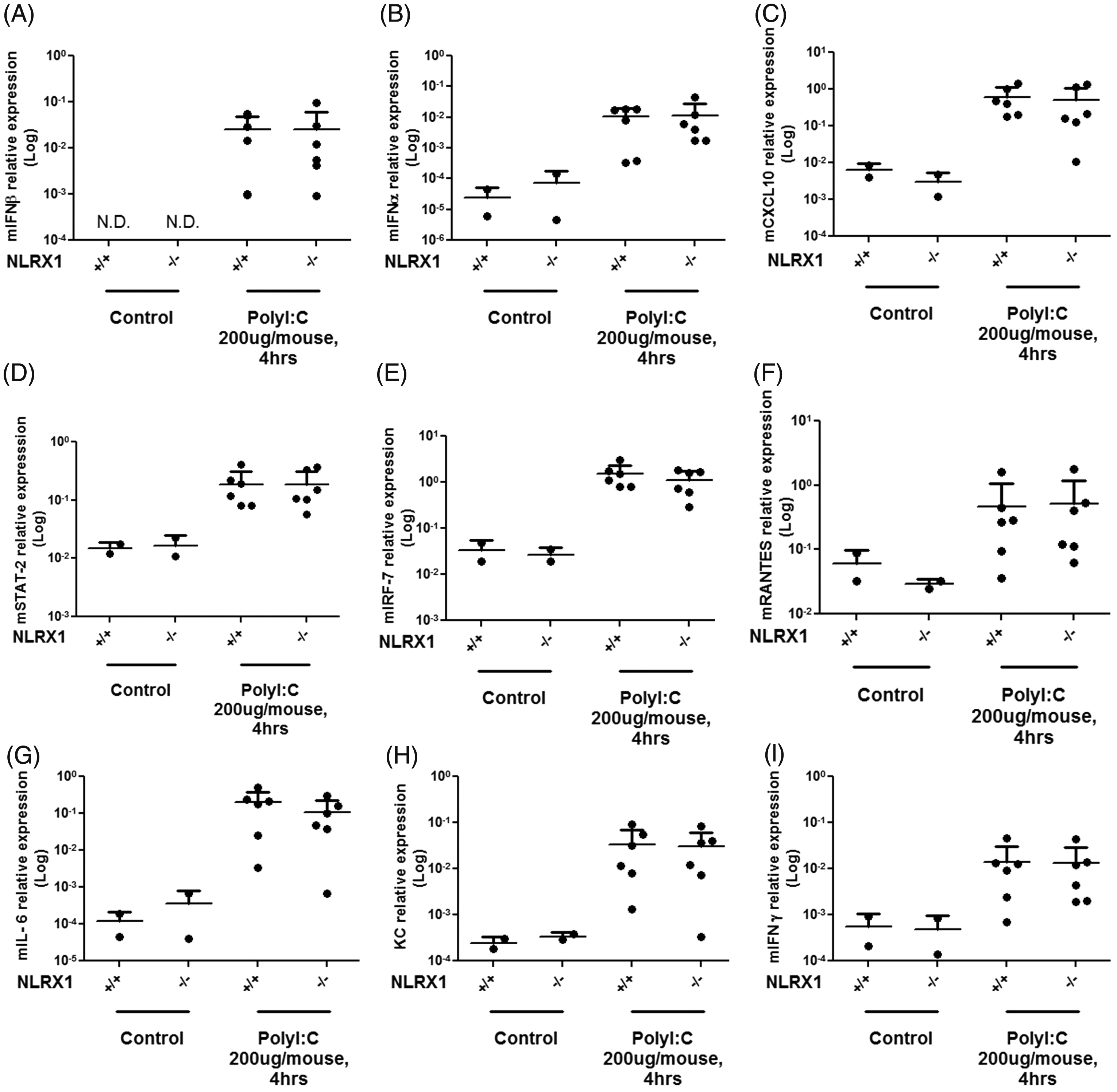
Together, these results strongly suggest that NLRX1-deficiency does not significantly affect early antiviral host responses to influenza A virus or Poly (I:C) in vivo.
Discussion
In this report, we present data supporting the conclusion that NLRX1 is not involved in MAVS-dependent antiviral signalling. Ex vivo experiments using NLRX1-deficient BMDMs and MEFs demonstrated that similar antiviral responses occurred during Sendai virus infection. In addition, NLRX1-deficient MEFs displayed normal IFN-β gene expression following EMCV or VSV infection, implying that MAVS-dependent antiviral signalling was not altered in these cells. Importantly, WT and NLRX1-deficient mice exhibited comparable levels of antiviral gene and cytokine expression following intranasal influenza A virus infection or i.p. injection of Poly (I:C).
These findings support the results described by the group of Dr J. Tschopp, who independently generated NLRX1-deficient mice and observed normal MAVS-dependent antiviral responses in NLRX1-deficient cells (MEFs and BMDMs) infected with Sendai virus, as well as in NLRX1-deficient mice injected i.v. with Poly (I:C). 8 In contrast with these observations, Allen et al. used another line of NLRX1-deficient mice and suggested that NLRX1 inhibited antiviral signalling through TRAF6, both in MEFs and in vivo, but surprisingly not in BMDMs. 13 These discrepant results could possibly be due to differences in these independently generated NLRX1-deficient mice. However, we demonstrated recently that human epithelial cells, silenced for NLRX1 expression and infected with Sendai virus, displayed normal induction of MAVS-dependent antiviral responses, 20 which further supports the conclusion that NLRX1 is not involved directly in negative regulation of antiviral signalling. Moreover, the proposed differential role played by NLRX1 in MEFs and BMDMs in antiviral responses 13 seems incompatible with the modulation of MAVS, as this signalling axis was shown to be indispensable for antiviral response to cytosolic viruses in these two cell populations. 21
Evidence supporting the negative role of NLRX1 in MAVS-dependent antiviral signalling relied on the fact that overexpression of NLRX1 resulted in dramatic inhibition of luciferase reporter assays dependent on interferon stimulated response element (ISRE) or NF-κB response elements, in virus-stimulated or MAVS-overexpressing cells.4,13,14 However, we also noted recently that this effect resulted from the non-specific inhibition of luciferase reporter assays at a post-transcriptional level (therefore not dependent on either ISRE or NF-κB response elements) in NLRX1-overexpressing cells. 20 Therefore, neither the results presented in the present study nor the data relying on NLRX1 overexpression in luciferase reporter assays support the conclusion that NLRX1 participates in the negative regulation of MAVS-dependent antiviral responses.
In conclusion, we were unable to confirm the observation that NLRX1 inhibits MAVS-dependent host antiviral responses in NLRX1-knockout MEFS and BMDMs, and in vivo after intranasal influenza A virus infection or i.p. injection of Poly (I:C). Further work is required to understand the function of NLRX1.
Footnotes
*These authors contributed equally to this work.
Funding
This work was supported by grants from Canadian Institutes of Health Research (CIHR) to D.J.P and Burroughs Wellcome Fund to S.E.G. F.S was supported by a Banting and Best Graduate Scholarship from CIHR.