
Abstract
Although rhinoviral infections, a major cause of asthma exacerbations, occur predominantly in upper airway bronchial epithelial cells, monocytic-lineage cells are implicated in establishing the inflammatory microenvironment observed during the disease. Human rhinovirus (HRV) is unique in that nearly genetically identical viruses bind either the ICAM-1 or low-density lipoprotein receptor (LDL-R). Within minutes of binding, HRV is capable of eliciting a signaling response in both epithelial cells and monocyte-derived macrophages. It is unclear whether this signaling response is important to the subsequent release of inflammatory mediators, particularly in cells not capable of supporting viral replication. We show here that the small molecular mass G-protein Rac is activated following exposure of macrophages to HRV serotypes known to be ICAM-1- and LDL-R-tropic. We demonstrate that inhibiting Rac resulted in the upregulation of TLR3 in macrophages exposed to major- and minor-group HRV, and resulted in increased release of IFN-α. Furthermore, inhibiting Rac in HRV-exposed macrophages attenuated activation of the stress kinase p38 and release of the pro-inflammatory cytokine CCL2, but inhibiting Rac did not affect release of the pro-inflammatory cytokine CCL5. These findings suggest that Rac is an important regulator in establishing the inflammatory microenvironment that is initiated in the human airway upon exposure to rhinovirus.
Introduction
Human rhinovirus (HRV) is responsible for both the majority of common cold infections and the majority of asthma exacerbations. Although the association of HRV with asthma exacerbation has been well documented, the molecular basis for HRV-induced asthma exacerbation is not well understood. 1
Within the lower airway, HRV contacts epithelial cells and alveolar macrophages, the predominant immune cells present in the lung. Both of these cell types possess receptors for HRV and both are capable of pro-inflammatory signaling.1–3 Although HRV infects epithelial cells productively, 4 macrophages are not a site of productive HRV replication.5,6 However, macrophages are important in establishing a pro-inflammatory microenvironment in the lung.5,7–9 Previous studies link HRV receptor-mediated signal transduction in macrophages to a number of biological endpoints associated with inflammation, including activation of inflammation-associated transcription factors, such as NF-κB,5,10–13 release of inflammatory cytokines8,14,15 and the dampening of the macrophage response to bacteria.16,17
HRV is a member of the virus family picornaviridae, a group of positive-sense RNA viruses with naked icosahedral capsids measuring approximately 20–30 nm in diameter. Other salient members of the picornaviridae family include enteroviruses, such as poliovirus and the Coxsackie viruses. HRV serotypes are divided into three groups—HRVA, HRVB and HRVC—based on sequence comparison.18,19 However, HRV serotypes can also be subdivided into groups based on which of two cellular receptors a particular HRV serotype will bind. Major-group HRV binds the ICAM-1 receptor,8,20,21 whereas minor-group HRV binds the low-density lipoprotein receptor (LDL-R). 22 Importantly, early signaling events associated with HRV receptor-mediated signal transduction for both receptors are not yet well characterized.
The Ras superfamily of low molecular mass G-proteins are important early participants in receptor-mediated signal transduction and many pathogens, including several viruses, have developed specialized mechanisms targeting GTPase function. The GTPases Cdc42, Rho and Rac, which belong to the Rho GTPase group within the Ras superfamily, contribute to the regulation of innate immune cell responses by modulating pathogen recognition, pathogen uptake and downstream release of pro-inflammatory mediators.23–26
The innate immune response plays an essential role in recognizing microbial motifs through a panel of pattern recognition receptors (PRRs). A wide variety of PRRs, including MDA-5, RIG-I, PKR, TLR3, TLR7 and TLR8, are known to play a role in the antiviral response.27–34 Of these, PRRs that recognize dsRNA, such as MDA-5, PKR and TLR3, are known to be important in sensing HRV in epithelial cells.35–38 TLR3 is able to signal independently of MyD88, leading to enhanceosome28,39,40 activation, increased expression of TLR3 and induction of type I IFNs, i.e. IFN-α and IFN-β.41–43 Indeed, TLR3 activation via rhinovirus exposure was described in epithelial cells and peripheral blood mononuclear cells exposed to rhinovirus as specifically leading to type I IFN production.35,37,44,45
In this study, we present evidence that following exposure of macrophages to HRV serotypes known to be ICAM-1- and LDL-R-tropic, Rac activation is observed and is part of a pro-inflammatory signaling cascade leading to mitogen-activated protein kinase (MAPK) p38 activation and chemokine ligand 2 (CCL2), monocyte chemotactic protein 1 (MCP1) release, but not the release of CCL5 (RANTES). We report the first demonstration of an important functional role for active Rac in suppressing the expression of TLR3 and IFN-α during HRV attachment to macrophages. These results provide a fuller understanding of the pro-inflammatory signaling milieu initiated during HRV-induced asthma exacerbation and reinforce the importance of certain G-proteins in initiating this pro-inflammatory environment in leukocytes.
Materials and methods
Reagents
For monocyte preparation, Lymphocyte Separation Media and Hanks buffered salt solution (HBSS) were purchased from CellGro (Manassas, VA, USA) and ammonium chloride potassium (ACK) lysing buffer from BioWhittaker (Walkersville, MD). Sigma Chemical Company (St Louis, MO, USA) was the source for protease inhibitor cocktail, phorbol myristate acetate (PMA) and platelet derived growth factor (PDGF). The Rac inhibitor NSC32766 was obtained from Tocris (Ellisville, MO,USA). Immunoblotting reagents were purchased from a variety of suppliers, including Santa Cruz Biotechnology (Santa Cruz, CA, USA; HRP-conjugated goat anti-rabbit IgG, anti-Rac antisera, Grb2 antisera and anti-Rho antisera), Thermo Fischer Scientific (Rockford, IL, USA; Supersignal™ chemiluminescence substrate reagents and immobilized glutathione agarose beads), R&D Systems (Minneapolis, MN, USA; anti-MCP-1 and anti-RANTES Ab pairs and IFN-α ELISA plate) and Cell Signaling (Beverly, MA, USA; anti-phospho-p38 MAPK antisera). Anti-CD14 and anti-CD86 was purchased from Becton Dickinson (San Jose, CA, USA). The reactive oxygen species (ROS) visualization reagent 2′-7′-dichlorodihydrofluorescein diacetate (DCFDA), dihydroethidium and propidium iodide for cell staining were obtained from Invitrogen (Carlsbad, CA, USA).
Isolation and purification/maturation of human blood monocyte-lineage cells
The protocol used for collecting blood samples was approved by the Lawrence University Institutional Review Board. Human blood samples were collected from healthy individuals, as described previously. 8 Whole blood was separated by density gradient, and leukocytes were collected from the buffy coat interface between the plasma and erythrocyte layers, as described previously. 8 Collected cells were distributed to 12-well tissue culture plates and cultured in RPMI 1640 (CellGro) containing 1% penicillin and streptomycin (Invitrogen) and 5% sterile-filtered, heat-inactivated human (type AB) serum. Adherent monocytes were matured for 7–10 d until a macrophage phenotype was achieved, as determined by flow cytometry. Purified human macrophages were lifted off the plate with Cell Dissolution Solution (Sigma) and the cell population was evaluated for CD14- and CD86-positive cells and viability (annexin V) by flow cytometry. Cell populations were typically 95% viable and 95% CD14-positive.
Cell culture
Human peripheral blood monocytes were cultured in RPMI 1640 (CellGro) with 5% human AB serum (CellGro, Manassas, VA) and 1% penicillin/streptomycin (Gibco, Grand Island, NY) at 37℃ in a humidified incubator with 5% CO2.
Preparation of HRV stock
HRV serotypes 16 and 1 A were gifts from the Jim Gern laboratory at the University of Wisconsin, Madison, WI, USA. Both serotypes are from the HRVA group. HRV was grown in HeLa cells and subsequently sedimented through a sucrose step gradient to remove exogenous protein and other contaminants. The HRV was then titered at 109 infectious particles/ml and stored at −80℃, as described previously. 8 RPMI 1640 enriched with human serum was used to prepare all necessary dilutions of both virus serotypes before virus was applied. Virus preparations were tested for endotoxin, as described previously, 8 and found to be endotoxin-free.
Pull-down assay for small molecular mass G-protein activation
BL21pLysS bacteria were transformed to express the appropriate glutathione-s-transferase (GST)-fusion proteins, and the proteins were harvested and stored as described previously. 46 The pull-down assay protocol was developed from one published previously. 46 Macrophages (1 million cells/well in a 12-well tissue culture plate) were exposed to HRV1A or HRV16 at a multiplicity of infection (MOI) of 10 in a time course. Cells were lysed in GST buffer and cell lysates were added to glutathione agarose beads bound to GST-fusion protein. GST-Pak was used for Rac pull-down assays and GST-Rock was used for Rho pull-down assays (both plasmids were gifts from Dr Paul Coffer). Beads were cleared by centrifugation and SDS-PAGE followed by immunoblot was performed.
SDS-PAGE and immunoblot analysis
Macrophages (1 million cells/well in a 12-well tissue culture plate) were exposed to HRV1A or HRV16 at a MOI of 10 in a time course. Selected wells were pretreated with 50 μM inhibitor NSC23766 30 min before virus treatment. Cell lysates were analyzed for MAPK activation using SDS-PAGE and immunoblot as described previously. 46 Rabbit primary Abs were used to probe for the presence of Rac, Rho, phospho-p38 and Grb2. Blots were visualized using HRP-coupled secondary Abs on a KODAK Image Station 4000 MM (Kodak, Rochester, NY) and Kodak MI Imaging Software (version 4.0.3).
Inhibitor time course for cytokine release
Inhibitor time courses for examining cytokine release took place over 24 h. The Rac inhibitor NSC23766 was applied to a final concentration of 50 µM 30 min before HRV was applied. 47 Both HRV16 and HRV 1 A were applied at a MOI of 10. Following the 24-h time course, the media supernatants were removed to cluster tubes and stored at −20℃ until ELISA could be performed.
ELISA
Following the inhibitor time course for cytokine release, the sandwich ELISA technique was used to probe for CCL2, IFN-α and CCL5 release. Half-size 96-well ELISA/immuno/assay (EIA) plates were coated at 4℃ for 8 h with coating buffer containing the concentrations of monoclonal capture Ab recommended by the manufacturer (R&D Systems). The plates were washed 3 times with 1× PBS with Tween 20 (PBS-T) to remove excess Ab and a standard curve of successive 1:2 dilutions of protein was prepared. The standards and the various samples were added to the 96-well plate in triplicate at 25 µl per well and the plate was incubated at 4℃ for 8 h. Monoclonal detection Ab was added to the plate at concentrations recommended by the manufacturer (R&D Systems) after washing 3 times in 1 × PBS-T and incubating the plate at 20–22℃ for 1 h. The plate was again washed 3 times in 1 × PBS-T. A 1:10,000 dilution of streptavidin-HRP was added to the wells and the plate was incubated for 20 min. The plate was then washed 3 times in 1 × PBS-T and 50 μl of 3,3′,5,5′-tetramethylbenzidine (TMB) component HRP-microwell substrate solution (BioFX, Owing Mills, MD, USA) was added to each well. When a blue color developed such that a gradation between standards could be detected visually, the reaction was stopped with 1 M hydrochloric acid. Optical density (absorbance) was read at 450 nm using a Molecular Devices (Sunnyvale, CA) ThermoMAX microplate reader with Spectrosoft software (version 6.2). Protein concentrations were calculated by averaging the triplicate values and interpolating from the standard curve. To determine whether differences between control and treated groups were significant, SPSS (version 16.0) was used to perform paired Student’s t-tests (alpha-reject = 0.05).
Flow cytometry measurements of ROS and TLR3
Cells were disassociated by trypsin and centrifuged for 2 min at 2000 g. The supernatant was removed and the pellet re-suspended in medium by gentle agitation at a concentration of approximately 1 million cells/ml and kept on ice until the experiment was performed. For reactive oxygen species (ROS) experiments, the cells were loaded with 5 μM DCFDA or dihydroethidium (DHE) 5 μM for 30 min at 37℃, transferred to flow cytometry tubes (500,000 cells/tube) and treated at 20–22℃ as described in the figure legends. For TLR3 experiments, the cells were fixed with 1% (wt/vol) paraformaldehyde. A total of 100,000 cells/ tube were stained with 1 mg/ml FITC-labeled anti-TLR3 Ab (cloneTLR3.7; eBioscience Inc., San Diego, CA) or FITC-labeled mouse IgG1 isotype control (BD Biosciences, Franklin Lakes, NJ) for 45 min. Flow cytometry was performed by measuring 10,000 cells on a BD Biosciences FACSCaliber flow cytometer. Propidium iodide-stained cells (dead cells) were not included in the analysis. Histogram statistics were analyzed by the program CellQuest (BD Biosciences, Franklin Lakes, NJ). Data were expressed as mean fluorescent intensity (MFI) of anti-TLR3-stained cells minus the MFI of isotype control-stained cells.
RNA extraction and quantitative RT-PCR
RNA was extracted from cells using the RNeasy method following the manufacturer’s instructions, including the optional DNase I digestion (Qiagen, Valencia, CA, USA) The Omniscript Reverse Transcriptase Kit (Qiagen) and oligo(dT)15 primer (IDT, Coralville, IA, USA) were used for cDNA synthesis, following the manufacturer’s instructions. Real-time PCR was performed using an ABI 7500 (Applied Biosystems, Foster City, CA, USA) using SYBR Green Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems). Primers for TLR3 and β-actin were purchased from Qiagen. The thermal cycler was set to perform an initial set-up (95℃, 10 min) and 40 cycles of denaturation (95℃, 15 sec) followed by annealing/extension (60℃, 1 min). After determining that all primer pairs used amplified with approximately equal efficiency, the relative amount of mRNA for the genes of interest was determined by subtracting the threshold cycle (Ct) values from the Ct value for the internal control gene β-actin. Data are depicted as fold difference from untreated control using the 2−ΔΔCt method.
Results
The small G-protein Rac is differentially activated in macrophages following HRV1A and HRV16 exposure
Because previous studies in the epithelial cell line A549 demonstrated that the small molecular mass G-protein Rho is activated in response to HRV16,
26
we sought to determine whether Rho would be similarly activated in macrophages. Although treatment of macrophages with either HRV1A or HRV16 did not produce Rho activation, as assessed by pull-down assay (Figure 1A, B), activation of the related small G-protein Rac was produced. Pull-down assays demonstrated Rac activation in macrophages following exposure to HRV1A or HRV16 at a MOI of 10 (Figure 1C, D). The activation of Rac occurred with differential kinetics that depended upon which HRV serotype provided the stimulus. In response to HRV1A exposure (LDL-R tropic), macrophages experienced escalating Rac activation from 2 min through 10 min followed by declining activation through the 30-min time point (Figure 1C). In response to HRV16 exposure (ICAM-1 tropic), Rac activation peaked at the 10 min time point and was sustained through the 30-min time course (Figure 1D). This is the first demonstration of HRV-mediated Rac activation in macrophages, suggesting a further line of investigation.
Rac and Rho activity following macrophage exposure to HRV1A and HRV16 in a 30-min time course. Monocyte-derived macrophages (1 × 106 cells/ml) were treated with HRV16 or HRV1A at a multiplicity of infection (MOI) of 10. In treatments with Rac inhibitor, macrophages were incubated with 50 μM NSC23766 for 30 min before HRV treatment. Activated Rho (A, B) or Rac (C, D) was pulled down and assayed by SDS-PAGE and immunoblot (IB) according to the procedure described in the Materials and methods. Treatment with PDGF for 15 minutes was used as a positive control for Rho activation. Equal protein for each pull-down was ensured by using total Rho or Rac as an internal control and probing with the appropriate Ab. Each blot is representative of five independent experiments.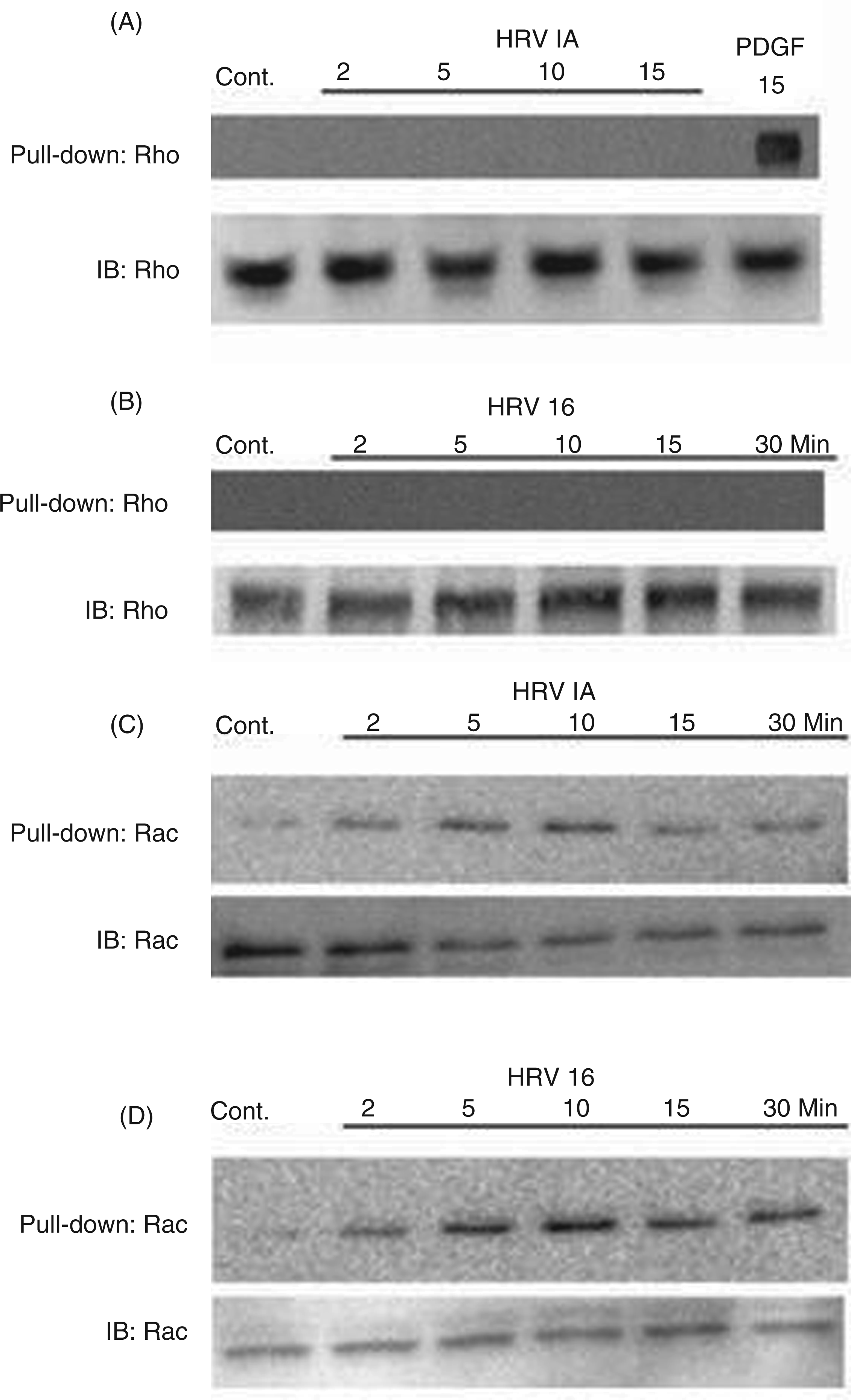
ROS are not elicited by exposure of macrophages to HRV16 or HRV1A
The production of reactive oxygen species is important for various macrophage functions and can be associated with Rac activation.23,48,49 Having observed Rac activation in rhinovirus-exposed macrophages, we next sought to establish whether HRV-stimulated Rac activation would lead to production of ROS in these cells. Macrophages were loaded with either the ROS-sensitive dye DCFDA or DHE. The addition of HRV16 or HRV1A at a MOI of 10 did not stimulate a detectable increase in ROS generation with DCFDA (Figure 2A) or DHE (Figure 2B).
Measurement of ROS production by monocyte-derived macrophages treated with HRV. (A) Monocyte-derived macrophages (1 × 106 cells/ml) were loaded with 5 μM DCFDA for 30 min at 37℃ followed by treatment of the cells with either vehicle (control), hydrogen peroxide, HRV16 at a MOI of 10 or HRV1A at a MOI of 10 at 20–22℃. The samples were then analyzed at the indicated times. (B) Monocyte-derived macrophages were incubated with 5 μM DHE followed by treatment with either vehicle (control), 10 ng/ml PMA, HRV16 at a MOI of 10 or HRV1A at a MOI of 10 at 20–22℃. The samples were then analyzed at the indicated times. The fluorescence of 10,000 cells was monitored by flow cytometry and is presented as relative linear fluorescence intensity. The data are representative of three independent experiments. RLMFI: relative linear mean fluorescence intensity.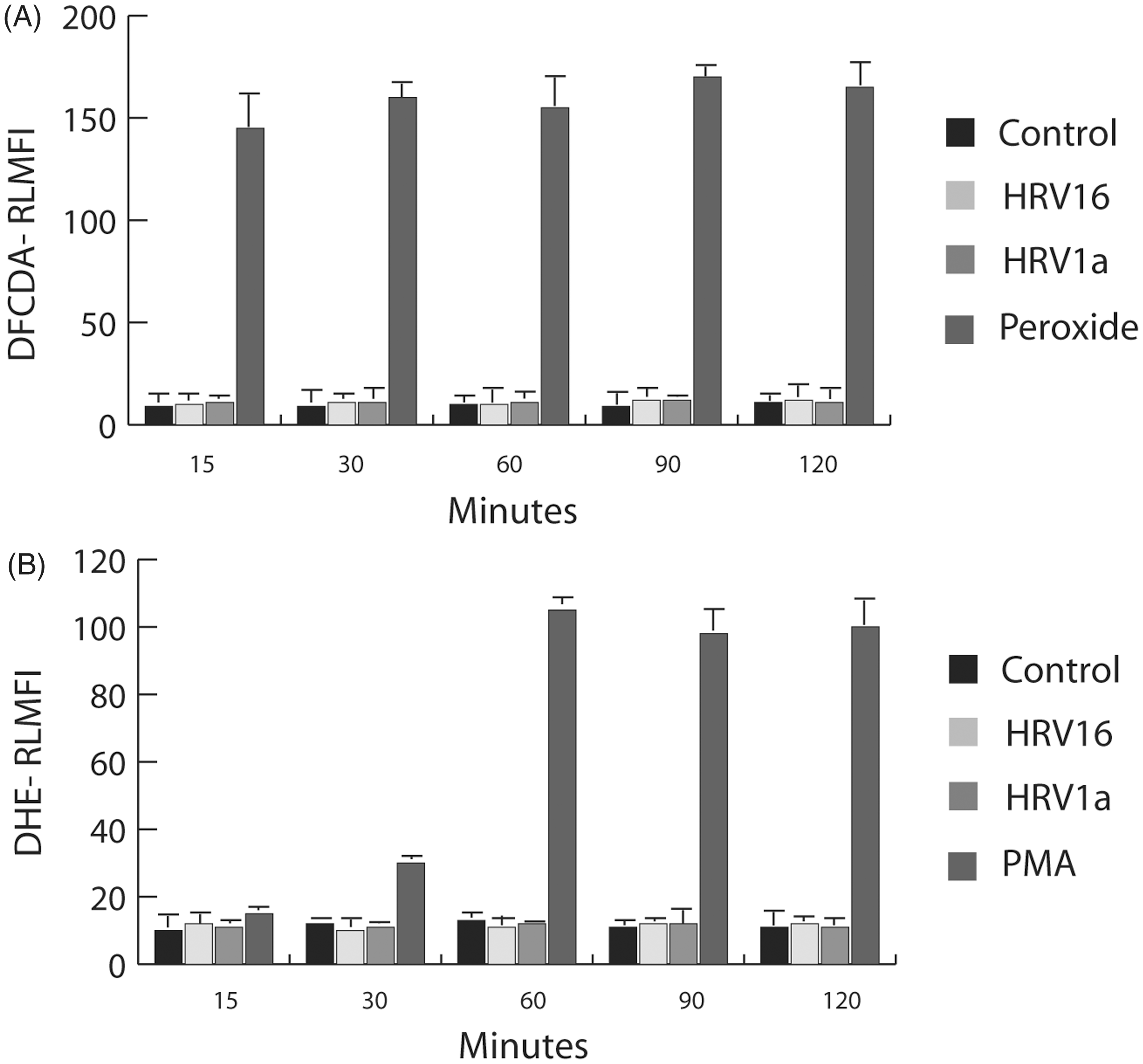
Pharmacological inhibition of Rac attenuates phosphorylation of stress-activated p38 MAPK following exposure of macrophages to HRV1A or HRV16
Previous studies have described the important role played by p38 MAPK in pro-inflammatory signaling cascades involving activation of Rac, and leading to phosphorylation of the transcription factor ATF-2 and the release of the pro-inflammatory cytokine CCL2 (MCP-1).8,50 Using NSC23766, a pharmacological inhibitor that blocks Rac activation by inhibiting the interaction between Rac and its guanine nucleotide exchange factors,
47
we demonstrated a decreased activation of p38 in macrophages responding to either HRV16 or HRV1A exposure at a MOI of 10 (Figure 3A, B). Although p38 activation escalated over a 90-min time course when macrophages were exposed to either HRV16 or HRV1A, adding NSC23766 30 min before applying the virus decreased p38 activation at the 90-min time point. The effectiveness of blocking Rac indicates that Rac signaling is necessary for activation of the p38 pro-inflammatory cascade that is induced in macrophages stimulated by HRV.
Activation of the MAPK p38 in macrophages exposed to HRV1A and HRV16 in a 90-min time course. Monocyte-derived macrophages (1 × 106 cells/ml) were treated with HRV16 or HRV1A at a MOI of 10. Selected 90-min time points were treated with the Rac-specific inhibitor NSC23766 for 30 min before HRV treatment. Activation of p38 MAPK was assayed by SDS-PAGE and immunoblot (IB) using an anti-phospho-p38 Ab. Equal protein loading was ensured by using Grb2 as an internal control and probing with an anti-Grb2 Ab. The blot is representative of five independent experiments.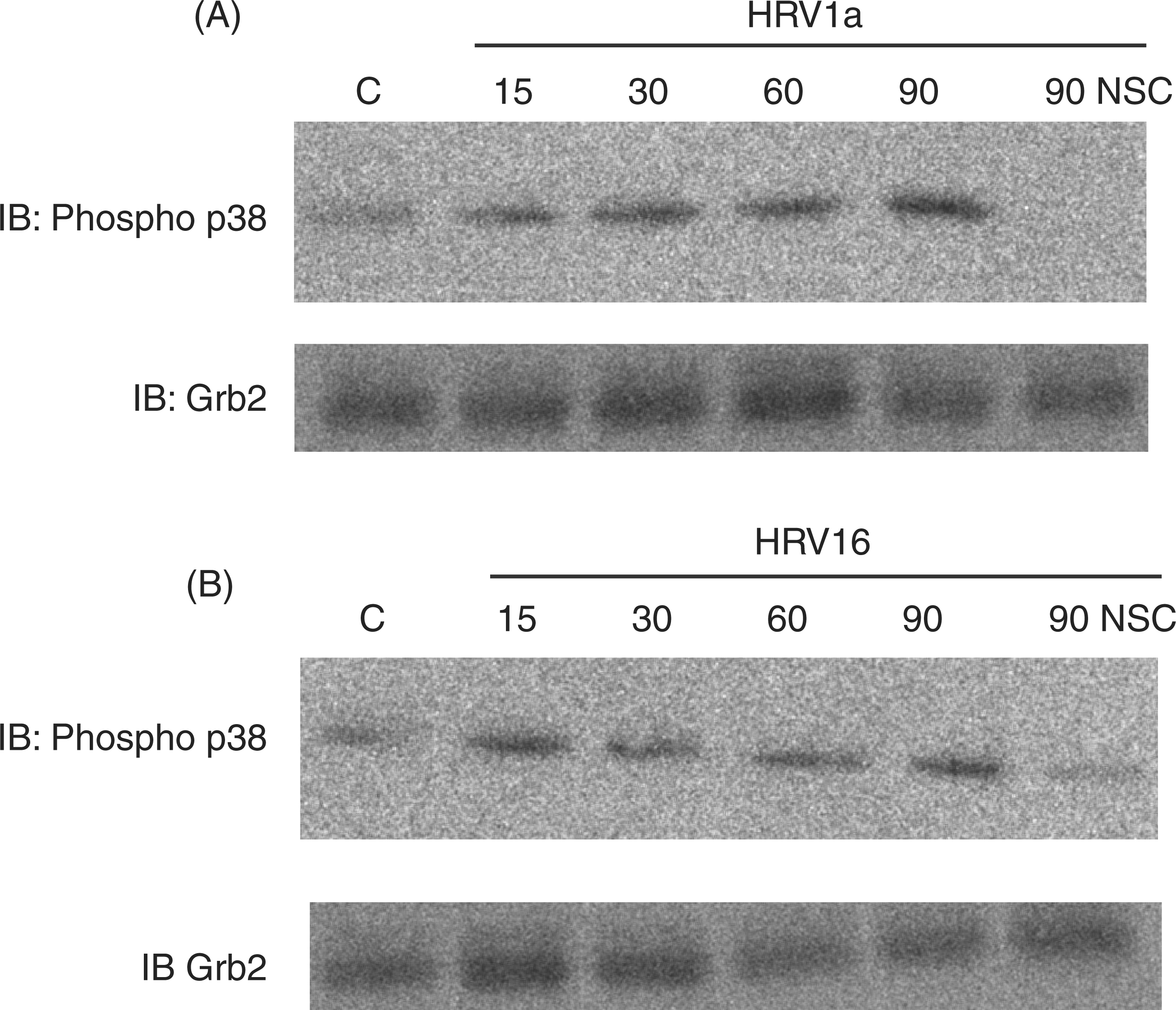
Inhibition of Rac affects the release of the pro-inflammatory cytokine CCL2/MCP-1 and IFN-α, but not the release of CCL5/RANTES
The production of many cytokines is associated with exposure of macrophages or epithelial cells to HRV.1,8,14,44,51–54 Because Rac inhibition led to attenuation of the p38 response in rhinovirus-exposed macrophages, we hypothesized that an attenuation of cytokine production might also be observed. Cytokines important in the asthmatic response, namely CCL2 (MCP-1), CCL5 (RANTES) and IFN-α, were obtained in cell supernatants and quantified via sandwich ELISA. NSC23766 inhibition of Rac in macrophages significantly downregulated the release of CCL2 in response to either HRV1 A or HRV16 exposure (Figure 4A) over a 24-h period. In contrast, the expression of IFN-α was significantly higher in macrophages treated with NSC23766 and exposed to either HRV16 or HRV1A (Figure 4C). However, inhibition of Rac by NSC23766 did not significantly alter the release of CCL5 (Figure 4B). These data demonstrate that the same cytokines are released by macrophages in response to major- and minor-group virus despite the fact that the two viruses bind to different receptors. Furthermore, we provide evidence that Rac activation is involved in the suppression of IFN-α production in HRV-exposed macrophages.
Release of the inflammatory cytokine CCL2, CCL5 and IFN-α by macrophages following 24 h of exposure to HRV1A and HRV16. Monocyte-derived macrophages (1 × 106 cells/ml) were treated with HRV16 or HRV1A at a MOI of 10. In treatments with Rac inhibitor, macrophages were incubated with 50 μM NSC23766 for 30 min before HRV treatment. After 24 h, cell supernatants were analyzed by sandwich ELISA, as detailed in the Materials and methods. (A) Release of the inflammatory cytokine CCL2 by macrophages. (B) Release of the inflammatory cytokine CCL5 by macrophages. (C) Release of the inflammatory cytokine IFN-α by macrophages. Presented here are the data from eight individuals and, in each of these eight experiments, triplicates were used to derive a mean. The data were subsequently combined and t-tests were performed for each analyte. The data were analyzed by paired Student’s t-tests with an initial alpha-reject value of 0.05. *P < 0.05.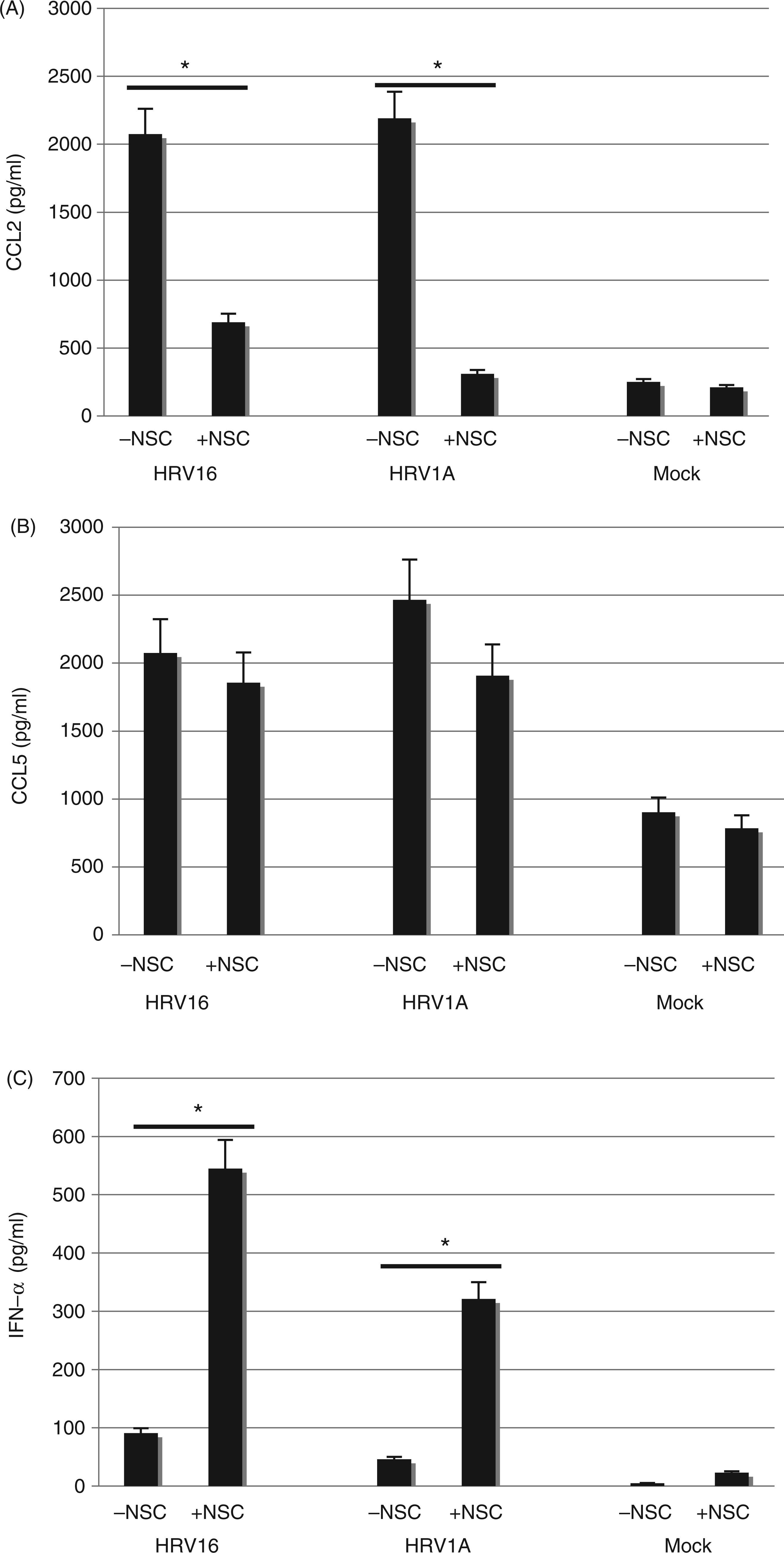
TLR3 expression is altered through inhibition of Rac
Because Rac inhibition led to an upregulation of IFN-α in rhinovirus-exposed macrophages, we sought to determine whether Rac inhibition positively regulated upstream constituents of the type I interferon signaling cascade, such as TLR3. Previous work in human epithelial cells has shown that TLR3 mRNA and protein expression increases 24 h after HRV infection.35,37,38,41,45,55 Macrophages exposed to NSC23766, HRV16 or HRV1A alone do not exhibit a statistically significant increase in TLR3 mRNA or protein expression when compared with mock-treated cells (Figure 5A, B). However, inhibition of Rac in HRV16 or HRV1A exposed cells results in a significant increase in TLR3 mRNA and protein expression (Figure 5A, B), suggesting that activation of Rac plays a role in regulating TLR3 production.
Expression of the TLR3 protein and mRNA by macrophages following 24 h of exposure to HRV1A and HRV16. Monocyte-derived macrophages (1 × 106 cells/ml) were treated with HRV16 or HRV1A at a MOI of 10. In treatments with Rac inhibitor, macrophages were incubated with 50 μM NSC23766 for 30 min before HRV treatment. (A) Expression of TLR3 protein was analyzed by flow cytometry as detailed in the Materials and methods. The fluorescence of 10,000 cells was monitored by flow cytometry and is presented as mean fluorescent intensity (MFI) of the TLR3 Ab minus MFI of the isotype control. The data are representative of three independent experiments. (B) Expression of TLR3 was assayed in blood monocyte-derived macrophages by qRT-PCR as detailed in the Materials and methods. The data are normalized to expression of the housekeeping gene β-actin and are expressed as gene expression fold change from untreated control. Error bars represent standard error (n = 5).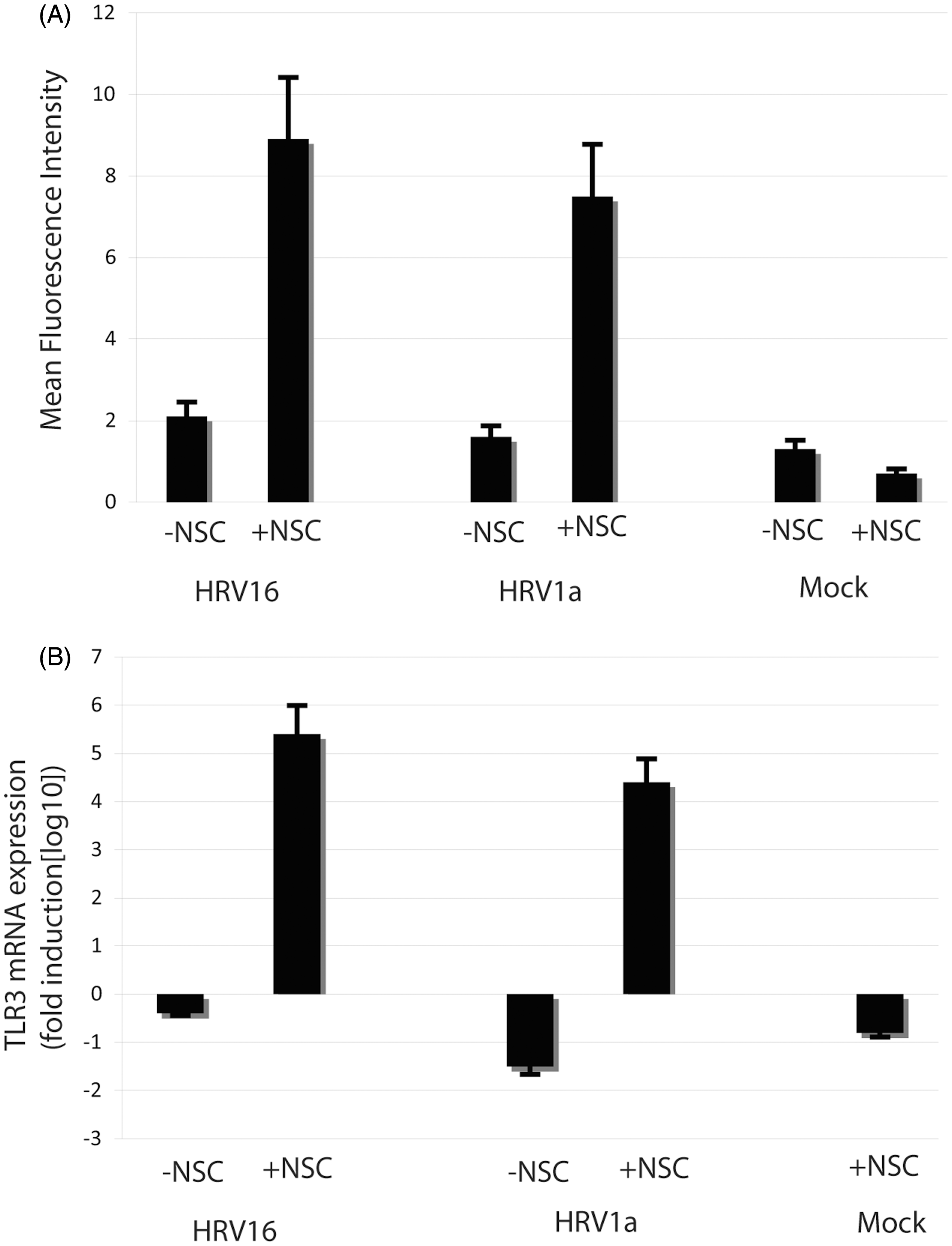
Discussion
Epithelial cells are the most studied cells involved in the release of HRV-induced cytokines; however, a number of studies indicate that the macrophage is also important in HRV infection.6,7,16,17,42,56,57 Although macrophages are not infected productively with HRV,5,6 they are known to express the HRV receptors ICAM-1 and LDL-R, and to release inflammatory cytokines, including CCL2, 8 various interferons 44 and CXCL10 (IP-10)14,56 in response to HRV exposure. Furthermore, several reports have demonstrated that monocyte-derived macrophages and alveolar macrophages behave identically to HRV exposure in signaling and cytokine secretion.8,56,58,59 We therefore sought to examine the initial signaling processes associated with the exposure of major- and minor-group rhinovirus to their respective ICAM-1 or LDL-R receptors on human monocyte-derived macrophages.
Recent studies have shown that the Ras superfamily small G-proteins play roles in viral replication60–62 and in the inflammatory response,23,63 and studies specific to HRV demonstrate that the small G-protein RhoA is important to HRV-induced cell signaling in epithelial cells. 26 Therefore, it is not surprising that HRV exposure stimulated the activation of the small molecular mass G-protein Rac and the stress kinase p38 (Figures 1 and 3), although ROS were not produced (Figure 2), as can occur in some cases of Rac activation. 63
Although the small molecular mass G-Protein Rac and the stress kinase p38 showed activation within 2 min and 15 min, respectively, in response to either HRV16 or HRV1A, the activation kinetics of the Rac-p38 signaling pathway appear to be different depending on whether major- or minor-group rhinovirus provides the stimulus. The differential kinetics imply receptor-specific differences in the rhinovirus-induced and Rac-mediated signaling pathway (Figures 1 and 3). This is the first study to observe a difference in signaling kinetics between major- and minor-group rhinovirus, suggesting an important area for further study.
The activation of p38, a MAPK, is associated with the production of asthma-related inflammatory cytokines, such as CCL2 (MCP-1), in macrophages exposed to HRV.8,14,56 Notably, pharmacological inhibition of Rac suppressed pro-inflammatory signaling involving phosphorylation of the MAPK p38 (Figure 3A, B). Consistent with this previous work on p38, we determined that pharmacological inhibition of the small G-protein Rac attenuated the release of CCL2 (Figure 4A) for both major- and minor-group HRV. These results are the first to implicate Rac in the pro-inflammatory signaling pathways initiated in macrophages upon exposure to HRV.
Similar to CCL2, CCL5 (RANTES) is a key inflammatory mediator for asthma released by epithelial cells during HRV infection.64,65 We report that CCL5 is also released by macrophages during HRV exposure, although pharmacological inhibition of Rac was not successful in blocking CCL5 release (Figure 4B). The differential effects of Rac inhibition on the release of the two inflammatory cytokines CCL2 and CCL5 suggest the presence of important differences in the mechanism by which HRV receptor-mediated signaling is transduced to these clinically-relevant biological endpoints.
The insensitivity of CCL5 release to Rac activation may be related to the effects of Rac signaling on TLR3 regulation.32,40,41 TLR3 is the PRR responsible for detecting dsRNA, and this receptor plays a critical role in the host antiviral defense through the production of pro-inflammatory cytokines and type I interferons. 32 Notably, epithelial cells upregulate TLR3 in response to challenge with either major- or minor-group HRV30,37,41,45,55 and this upregulation in response to virus is enhanced synergistically by many other potent asthma triggers, such as diesel exhaust 66 and cigarette smoke. 36 Therefore, we sought to determine whether HRV Rac-mediated signaling would also be important in regulating expression of TLR3 in macrophages.
Exposing macrophages to HRV following Rac inhibition led to a substantial upregulation of TLR3 protein and mRNA (Figure 5A) and a noticeable upregulation of IFN-α protein (Figure 4C). However, Rac inhibition without HRV stimulation led to no noticeable differences in TLR3 or IFN-α protein expression. Thus, whereas Rac inhibition has apparent anti-inflammatory properties in the context of the p38/CCL2 cascade, Rac inhibition may also have some paradoxic pro-inflammatory activity, in contrast with a previous report, enhancing the response of the TLR3/IFN-α cascade. 24
Excessive activation of a TLR-triggered response may enhance cancer progression 67 and induce inflammatory autoimmune diseases.39,68 Therefore, the tight regulation of TLR signaling pathways and control of the inflammatory response is important for maintaining homeostasis and avoiding excessive damage to host tissues.40,69,70 Within this context, Rac activation may serve as a negative regulatory component of the TLR3 and IFN-α expression pathways through the interferon regulatory factor (IRF) family of transcription factors, 71 although the mechanism remains unclear. Our findings suggest that HRV may exploit the endogenous negative regulation of the TLR3/IFN-α cascade in macrophages via receptor-mediated triggering of Rac activation.
The normal homeostatic mechanisms regulating the immune response to HRV in humans are not well understood. An increasing body of evidence suggests that airway macrophages from asthmatics have a reduced capacity for the synthesis of innate IFN-α compared with airway macrophages from healthy people.58,72,73 Furthermore, evidence suggests that IFN-α can modulate adaptive immunity by inhibiting Th2 commitment and secretion of Th2 cytokines while supporting Th1 and Th17 polarization and effector function.74,75 Thus, a further understanding of Rac signaling in the context of IFN–TLR–macrophage biology will likely provide insights into the mechanisms of inflammation and new approaches for rational therapeutic intervention during HRV infections and asthma exacerbations.
Footnotes
Acknowledgements
Paul J. Bertics was an inspiration for this work as a mentor and friend. He will be missed. We are grateful to Dana Raugi, Megan Wilson, Megan Frear and Bethany Kondiles for their contributions to the data presented here. We would also like to thank the Paul Bertics laboratory, Jim Gern, Yury Bochkov and Wai-Ming Lee at the University of Wisconsin-Madison for their contribution of viral stocks and reagents.
Funding
This work was supported by the Ronald E. McNair Postbaccalaureate Achievement Program, the Henry Merritt Wriston Scholarship, the Lawrence Excellent in Science Fund, NIH R15 AI065505-01A1 and NSF 0521112.