
Abstract
Traumatic brain injury (TBI) is common and potentially devastating. Traditional examination-based patient monitoring following TBI may be inadequate for frontline clinicians to reduce secondary brain injury through individualized therapy. Multimodal neurologic monitoring (MMM) offers great potential for detecting early injury and improving outcomes. By assessing cerebral oxygenation, autoregulation and metabolism, clinicians may be able to understand neurophysiology during acute brain injury, and offer therapies better suited to each patient and each stage of injury.
Hence, we offer this primer on brain tissue oxygen monitoring, pressure reactivity index monitoring and cerebral microdialysis. This narrative review serves as an introductory guide to the latest clinically-relevant evidence regarding key neuromonitoring techniques.
Keywords
Introduction
With 27 million new cases and 8.1 million years lived with disability in 2016, traumatic brain injury(TBI) represents the leading cause of global disability.1–3 In the intensive care unit (ICU), TBI treatment focuses on preventing and treating secondary brain injury. This has traditionally meant monitoring and mitigating physiologic derangements in blood pressure, oxygen saturation, the partial pressure of carbon dioxide (CO2), and intracranial pressure (ICP).
In isolation, the clinical examination may not be sufficient to detect events that have been associated with worse outcomes, such as elevations in ICP, reduced cerebral oxygen availability, or subclinical seizures. Moreover, many of the treatments used in TBI management may further confound the clinical examination, such as analgesics, sedation, and pharmacologic paralysis. Advanced cerebral monitoring tools may be used to detect evolving secondary injury before, rather than during or after, overt neurologic deterioration (Figure 1).4,5 Collectively, these tools are referred to as multimodality monitoring (MMM). Broadly, MMM involves the use of multiple techniques to measure and integrate information regarding cerebral blood flow (CBF), oxygenation(i.e., measuring the balance between delivery and use), ICP, autoregulation, metabolism, and cortical function. For this review, we will provide a practical primer with a focus on techniques for monitoring cerebral oxygenation, autoregulation, and metabolism.
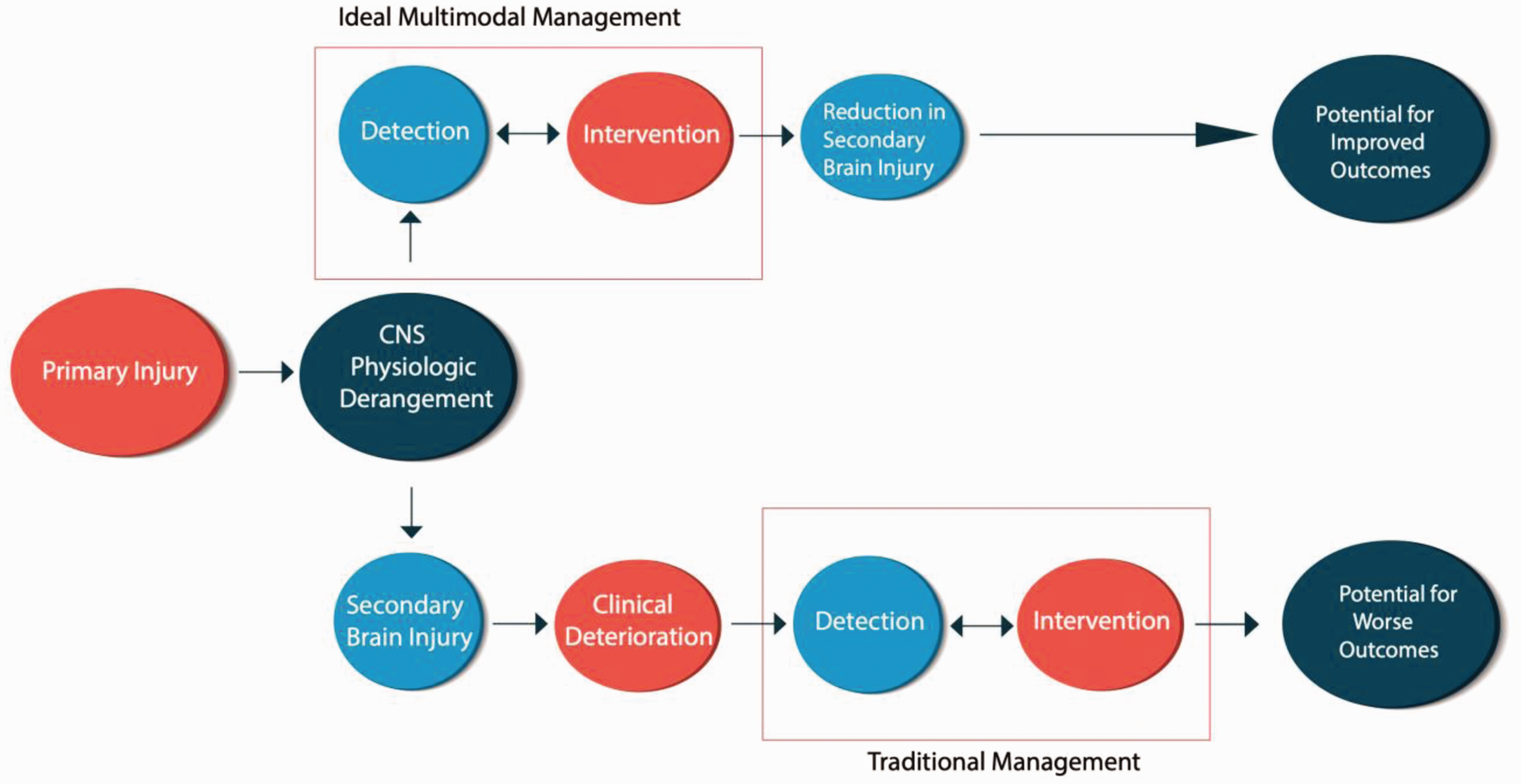
Hypothetical Model for the implementation of MMM in traumatic brain injury.
Cerebral oxygenation monitoring
For at least three decades, clinicians have had the technology to interrogate cerebral oxygen delivery and utilization. Available tools include jugular bulb mixed venous oximetry, near infrared spectroscopy, oxygen-15 positron emission tomography (PET) and brain tissue oxygen tension monitoring (PbTO2). This review focuses on PbTO2 monitoring, as it is extensively researched thus far and is being further evaluated in ongoing randomized controlled trials (RCTs).
PbTO2 monitors use a Clark electrode (Licox Integra Monitor, Integra Neurosciences, Plainsboro, NJ, USA), or an oxygen quenching method (Raumedic Neurovent-PTO, Raumedic, Münchberg, Germany), to determine the partial pressure of oxygen in brain parenchyma. The Clark electrode consists of a cathode and anode, surrounded by a thin membrane, through which oxygen diffuses from a 15 mm radius. Dissolved oxygen is reduced at the cathode, thereby allowing the sensor to estimate the partial pressure in the surrounding brain parenchyma. The 0.5 mm probe is usually placed in frontal lobe white matter and requires approximately 30 to 60 minutes for calibration before producing reliable readings. Catheters also require recalibration daily via O2 and CO2 reactivity testing. 6
When using a PbTO2 monitor, probe location matters, as it affects interpretation. Although still debated, monitors are usually placed in the unaffected hemisphere. The justification is that this enables detection and mitigation of cerebral hypoxemia in tissue that is comparatively healthy and salvageable. In contrast, proponents of placing the probe in (or near) damaged tissue argue that this could preserve at risk or “penumbral” tissue. The concern is these tissues may already be less responsive to therapy. 7 In the case of diffuse injury, the probe is usually placed in the non-dominant hemisphere for practical and safety reasons to minimize the risk of harm in the case of iatrogenic hemorrhage.
“Normal” PbTO2 values are usually >23 +/− 7 mmHg. 6 However, there is no universally accepted threshold at which tissue damage occurs. PbTO2 values of <15 mmHg are associated with a widening lactate/glucose ratio and increasing glycerol levels, both of which suggest cellular energetic failure.8,9 Consistent international consensus guidelines provide inconsistent direction on the most optimal PbTO2 target. Consequently, the neurocritical care society considers both 15 or 20 mmHg as acceptable, while the Brain Trauma Foundation (BTF) does not provide specific guidance in this area. 10 In on-going clinical trials such as BOOST III and BONANZA, the PbTO2 interventional threshold is 20 mmHg.11,12 Accordingly, many experts recommend a PbTO2 target >20 mmHg following TBI, however regional variation may be observed. 13
Following TBI, cerebral hypoxemia may be more common than previously thought. Using fluorine PET, TBI patients have demonstrated diffuse cerebral ischemia distant from traumatized tissue in seemingly healthy parenchyma. 14 Using the threshold of a PbTO2 value <20 mmHg, hypoxemia occurred in, at least, half of patients (50–86%) in the first few days post injury.15–17 Importantly, episodes occurred despite normal ICP or cerebral perfusion pressure (CPP), suggesting cerebral hypoxemia would be missed with traditional neuromonitoring techniques. 18 In short, cerebral hypoxemia would have been missed without MMM.
The presence of cerebral hypoxemia is important as it may be impactful to patient outcomes. Low PbTO2 values are associated with higher patient mortality,19–21 and the detrimental effect appears to be “dose related”, namely worsened by duration and degree. 18 In cohort studies, those TBI patients who spent more time with low PbTO2 had worse Glasgow Outcome Scores exams (GOSE) and lower functional status at 30 days and 6 months.16,20–22 Furthermore, patients who are not responsive to PbTO2 directed therapy- i.e. patients whose cerebral hypoxemia is irreversible- are less likely to survive. In a prospective observational cohort study, survivors had a 71% response rate to PbTO2 directed treatment compared to 44% in non-survivors. 23
While PbTO2 directed therapy shows promise for ameliorating cerebral hypoxemia, choosing which therapy to use is complex. There are four components to consider when treating a low PbTO2 value: oxygen delivery, diffusion, demand and utilization (Figure 2). This, in turn, means concomitantly addressing ICP, CPP, systemic oxygenation, hemoglobin, temperature, sedation, shivering, the partial pressure of carbon dioxide and seizures (See Figure 3). Pascual et al. found the majority of patients require only a single intervention to normalize PbTO2 values. However, no individual intervention will optimize all low PbTO2 values. 24 The SIBICC guidelines provide a tiered approach to managing patients with ICP and PbTO2 monitoring. 13 This strategy divides patients into four groups based on dichotomized ICP and PbTO2 variables. The groups are then labelled by type including Type A (normal ICP/PbTO2), Type B (elevated ICP but normal PbTO2), Type C (Normal ICP and low PbTO2) and Type D (elevated ICP and low PbTO2). 25 While large RCTs are on-going, the SIBICC guidelines provide level III evidence to guide and standardize PbTO2 therapy.
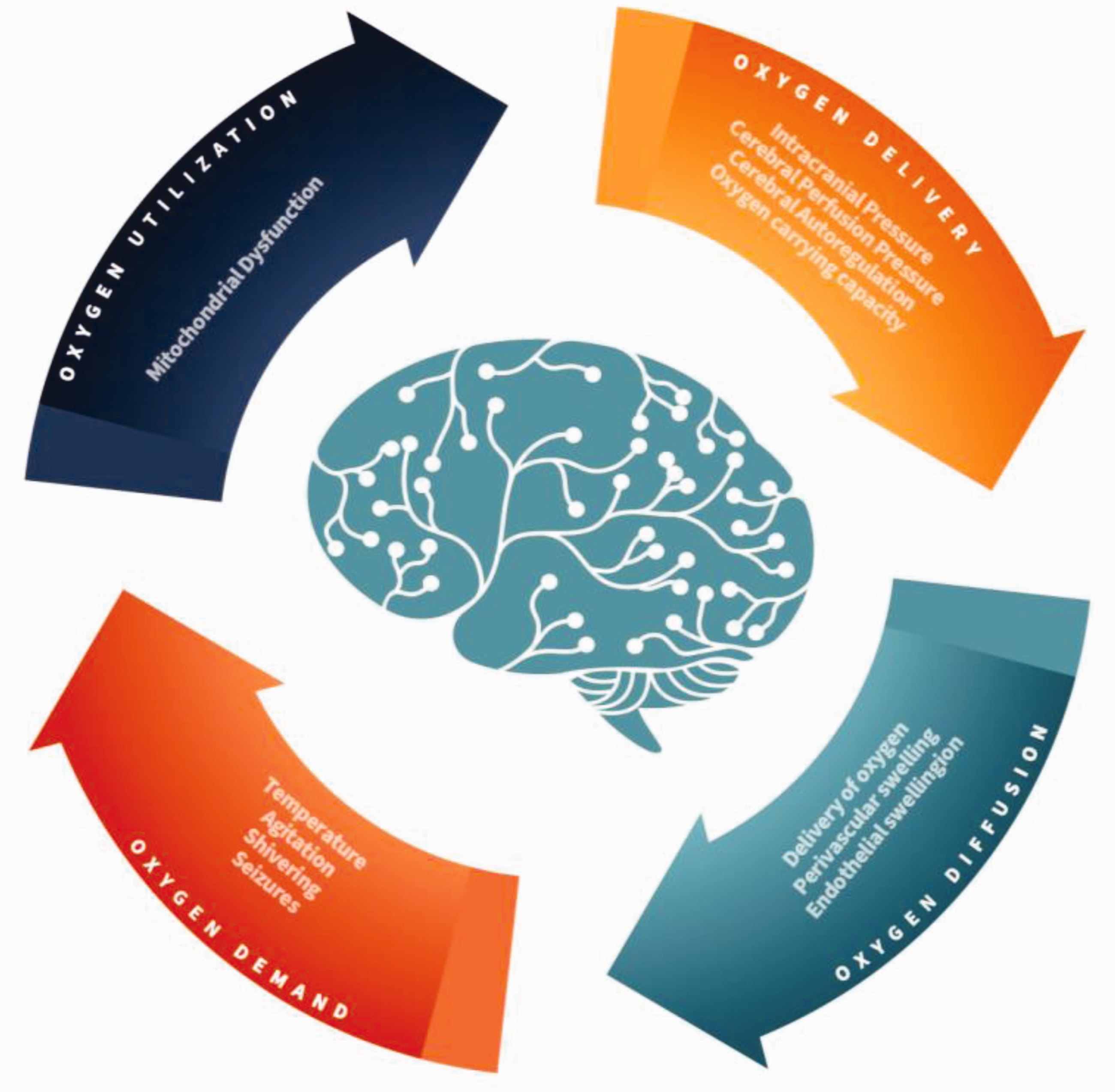
Determinants of cerebral oxygenation.
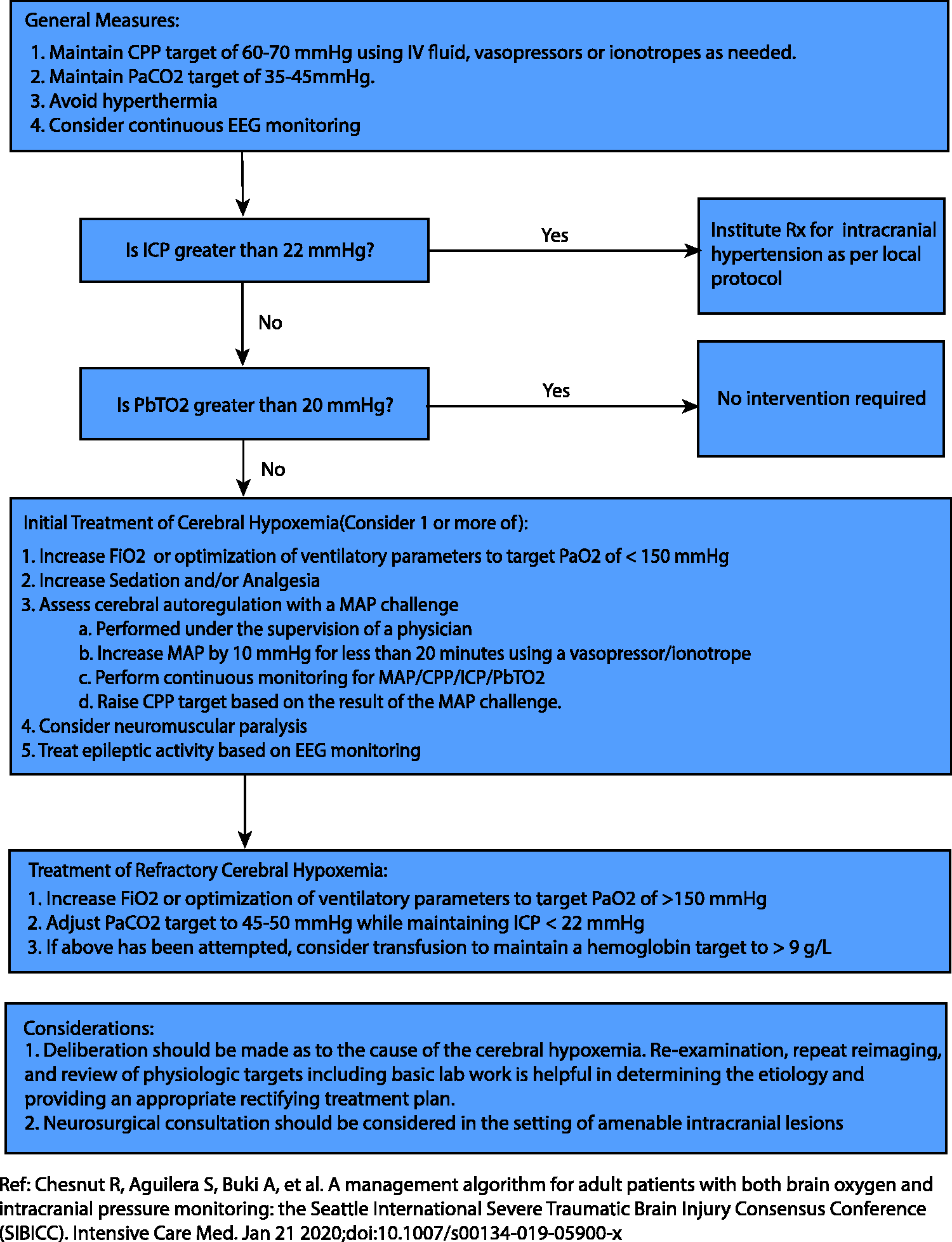
Algorithm for the management of cerebral hypoxemia.
Overall, there is evolving evidence to support monitoring brain tissue oxygenation, however RCTs are still needed to demonstrate treatment of PbTO2 positively impacts outcomes in TBI. BOOST-2 was a pilot RCT that demonstrated protocolized PbTO2 & ICP directed therapy compared to an ICP directed treatment alone was both feasible and safe. Specifically, those in the PbTO2 & ICP directed therapy arm spent less time with cerebral hypoxemia (PbTO2 < 20 mmHg) 26 compared to those in the ICP only group. Moreover, there was a non-significant trend towards improved outcomes (although, importantly, the BOOST-2 trial was not powered for this outcome). 26 As a result, clinicians are eagerly awaiting results from three RCTs using combined PbTO2-ICP directed therapy in severe TBI.11,12,27
Monitoring cerebral autoregulation
The first description of cerebrovascular reactivity is credited to Neils Lassen, who, in 1959, established that CBF remains nearly constant across a wide range of CPP. 28 Cerebral vascular reactivity is dependent on multiple contributors, including myogenic(e.g. changes in transmural pressure), neurogenic (e.g. acetylcholine, serotonin, etc.), endothelial (e.g. nitric oxide, thromboxane, etc.) and metabolic (e.g. pH, CO2, etc.) factors. 29 Changes in these variables causes cerebral arterial dilation or constriction which, when functional, maintains a constant CBF. The myogenic component, referred from here on as cerebral autoregulation, refers to the contraction or dilation of cerebral blood vessels in response to changes in transmural pressure. 29 When cerebral autoregulation is intact, cerebral arterioles constrict or dilate in response to high or low mean arterial pressure (MAP), respectively, which ensures a near-constant CBF across a wide range of CPP (Figure 4). In the setting of brain injury, cerebral autoregulation may become impaired. Autoregulatory exhaustion may predispose patients to periods of cerebral ischemia, secondary to inappropriately low CBF, or cerebral hyperemia, due to abnormally high CBF, even at guideline-recommended MAP, ICP, and CPP targets.30,31 Therefore, monitoring cerebral autoregulation allows clinicians to individualize CPP targets to avoid further secondary injury from cerebral ischemia or hyperemia.
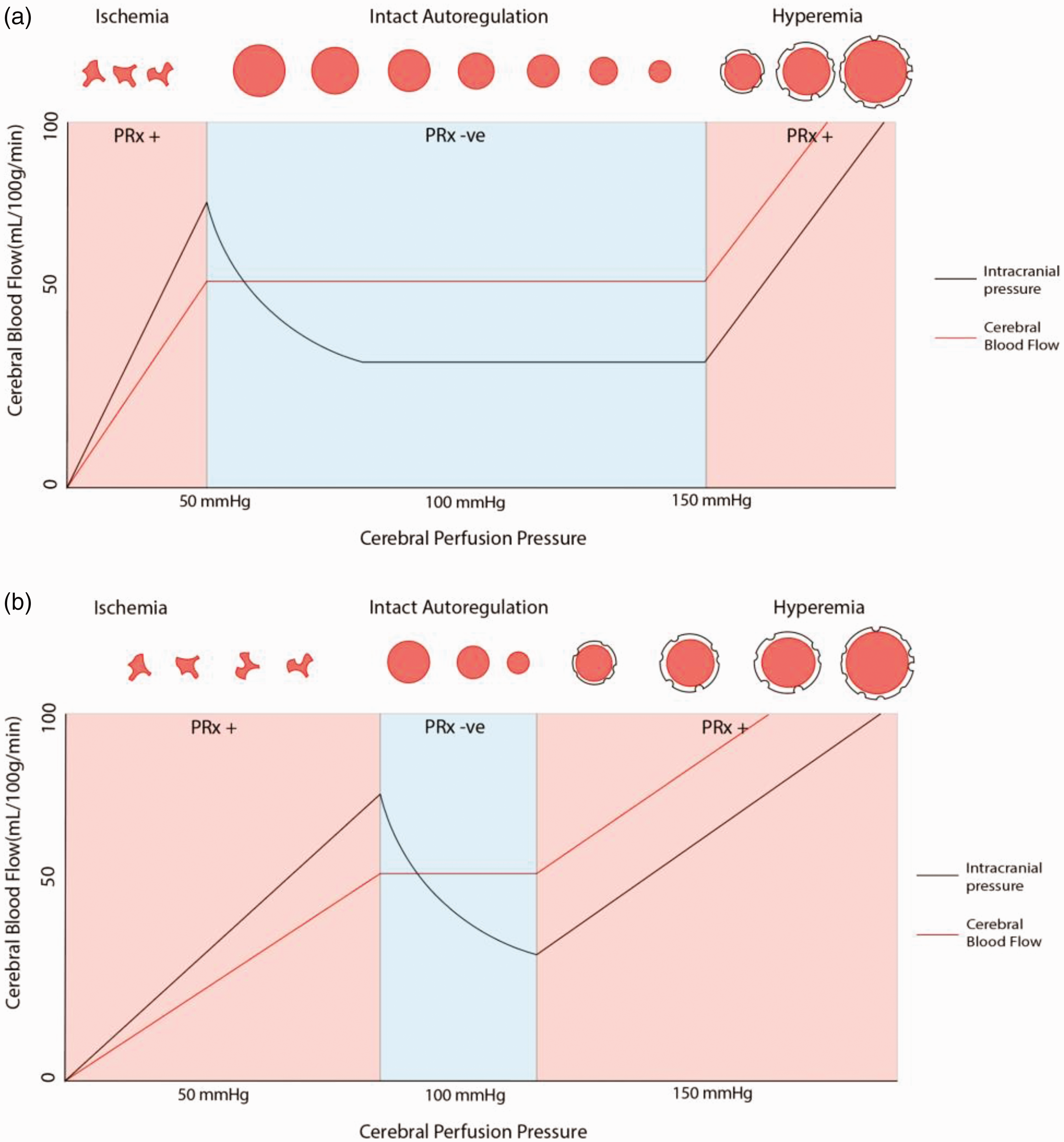
Relationships between CBF, CPP and ICP with intact and disturbed autoregulation.
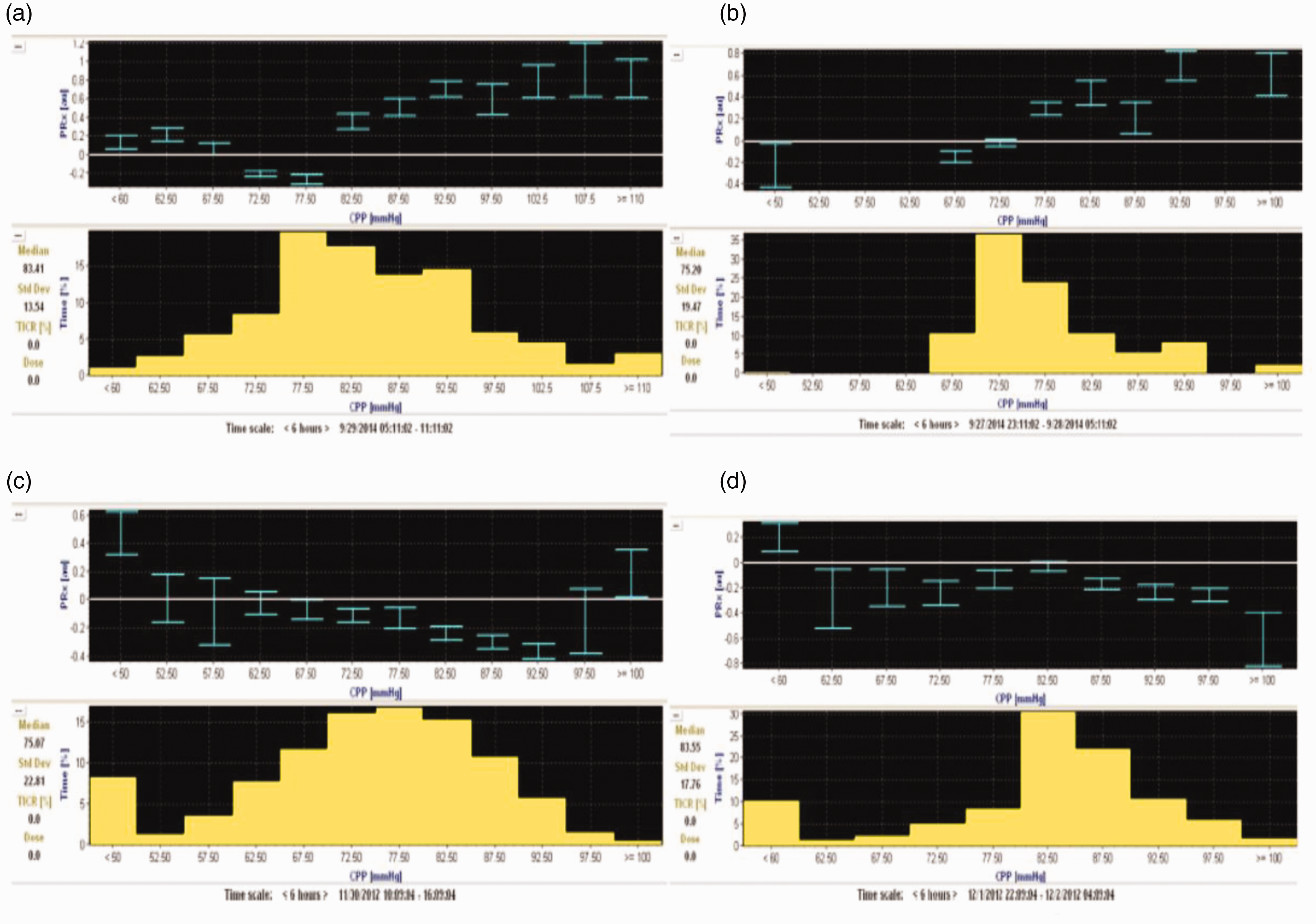
Common phenotypes of PRx Derived autoregulatory curves in severe TBI. (a) U-shaped curve phenotype where CPPopt is identified at 75-80 mmHg. (b) An Ascending curve demonstrating worsening autoregulatory status as CPP is increased, therefore the CPPopt is found at the lowest CPP with reasonable sampling 65-70 mmHg. (c) Descending curve pattern where autoregulatory function is optimal at a higher CPP than BTF guidelines (90-95 mmHg) (d) No clear relationship can be discerned by the CPP sample. Figure reproduced with the expressed written consent of Springer Publishing Group.
By measuring changes in ICP in relation to changes in CPP over time, a Pearson’s moving correlation coefficient can be used to mathematically quantify this relationship, known as the Pressure Reactivity Index (PRx), which represents the autoregulatory status of the patient. When cerebrovascular autoregulation is intact, rapid changes in transmural pressure from increased CPP cause cerebral arterial constriction, which leads to a reduction in cerebral blood volume and is detected by a subsequent decrease in ICP. Mathematically, this relationship is represented as a negative PRx value. Alternatively, if autoregulation is exhausted, increases in CPP cause passive arterial dilation rather than constriction as transmural pressure increases. Consequently, there is an increase in cerebral blood volume and is detected as an increase in ICP. Accordingly, this relationship is demonstrated as a positive PRx value. Autoregulatory failure is typically described by a PRx value of >0.2–0.3; however, failure is more likely a continuum between 0 and +0.35 rather than a binary threshold. 32 It is during this state clinicians should be particularly concerned about worsening secondary injury from ischemia or hyperemia as the brain may receive insufficient or excessive CBF in response to an ordinarily physiologic CPP. Because PRx is determined from ICP & MAP, it can be sampled with an extremely high frequency, providing minute to minute monitoring information and represents a continuous autoregulation monitoring method compared to a static method like transcranial doppler which represents only a single point estimate at the time of the study. Continuous monitoring of cerebral autoregulation using PRx provides a distinct advantage compared to static methods as changes in pH, sedation level, CO2, temperature and many other factors may impact cerebrovascular reactivity dynamically.
Aries et al. collected PRx values over time, averaged over five seconds (or longer) and graphed the PRx against the CPP for periods of four hours. 33 This method provides an estimate of the individual patient’s autoregulatory function across a spectrum of CPP values and the subsequent calculation of an individualized “optimal” target CPP (CPPopt), namely the CPP range with the lowest PRx value. 34 Software systems, such as ICM+(Cambridge, UK), can graph average PRx against CPP in real-time. For those without specialized software, cerebral autoregulatory status can be determined by observing the ICP response to a bedside blood pressure challenge. 35 Experts recommend increasing MAP by 10 mmHg, using vasopressors, while assessing the response in ICP for 10–20 minutes. Generally, one should not target a CPP of less than 50 or greater than 90 mmHg for concern of precipitating iatrogenic cerebral ischemia or hyperemia. 35
When using a PRx curve to determine CPPopt, four patterns are commonly observed; a U-shaped, ascending, descending, or absent curve (Figure 5). An ascending or descending autoregulatory profile suggests the clinician should consider a lower or higher MAP, respectively (Figure 6). Using four-hour epochs, CPPopt can be reliably determined in just over half (55%) of the recording time. 33 To more consistently determine CPPopt, a multi-windowing method has recently been validated. This generates a mean estimation of CPPopt based on data from multiple PRx calculation windows, using epochs of 2–8 hours. 36 , 37
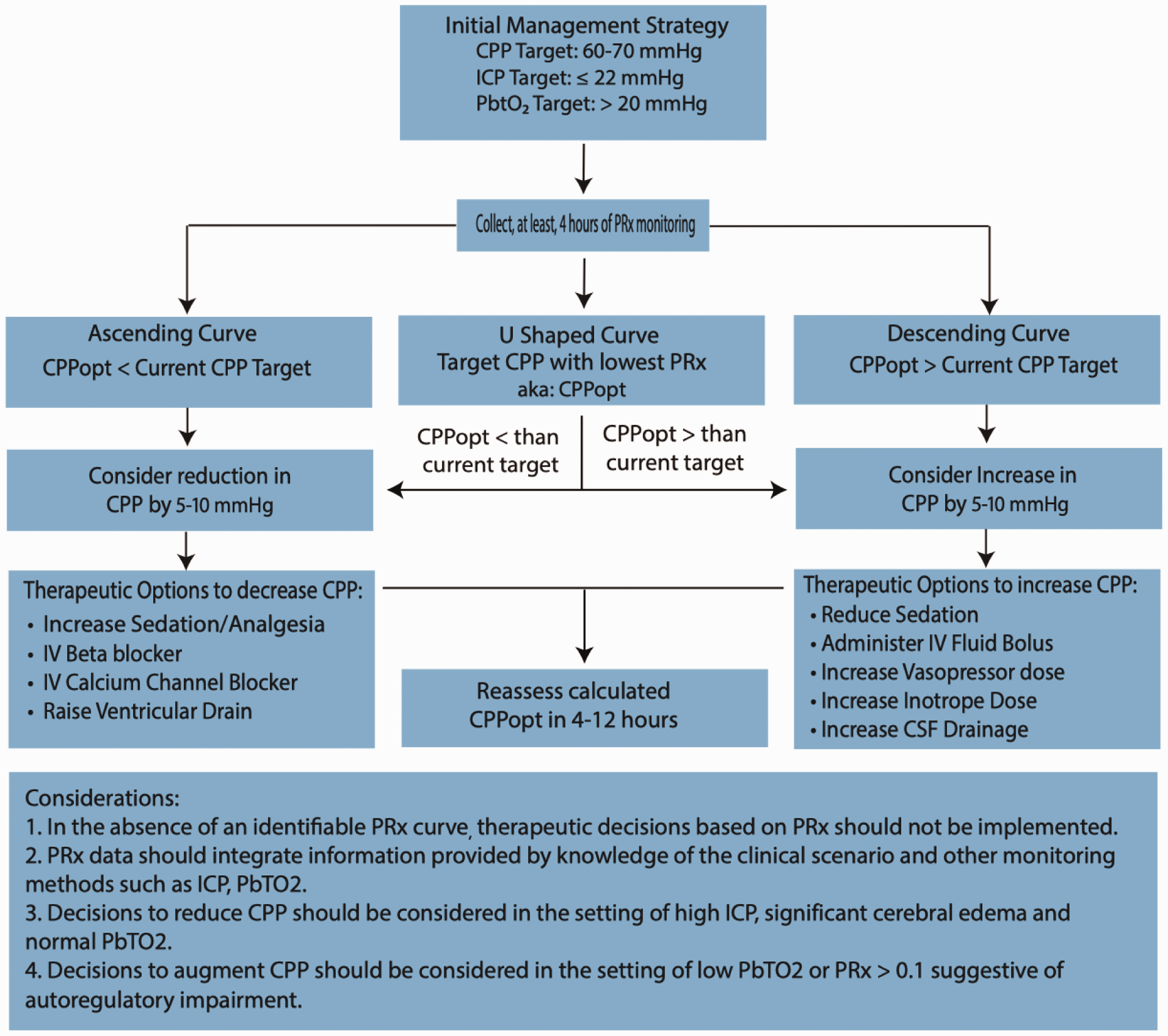
Proposed model for the use of PRx derived CPPopt in severe TBI.
Importantly, the validity of a PRx value at a target CPP is dependent on how long the patient spent in that particular CPP range. If a patient spent a small proportion(e.g. <5%) of monitored time at a particular CPP, then clinicians may choose to downplay the usefulness of that PRx value, because of insufficient sampling. The period of time required to collect a “definitively valid” sample is unclear, however, it is obvious that longer sampling times provide a more reliable understanding of a patient’s autoregulatory status at that CPP.
International guidelines have largely not acknowledged the inter-individual differences in autoregulation or its importance at the time of this writing. The BTF guidelines recommend a CPP of 60–70mmHg after TBI, but acknowledge this may fail to account for patient differences. 38 Regardless, deviation from individualized CPPopt appears to correlate with outcome. Those patients who spent time below CPPopt or had a more significant discrepancy between CPPopt and CPPactual, had worse neurologic outcomes, particularly if they had disturbed autoregulation. 39 Therefore, it is possible deviation from CPPopt may worsen secondary brain injury by the previously mentioned mechanisms. Work by Mathieu et al. supports this hypothesis. TBI patients who spent more time with a PRx > 0.25 had worse cerebral edema on imaging. 40 Because patients with disturbed autoregulation appear to be at higher risk of secondary brain injury, PRx can help predict neurologic outcome following TBI. 41
Despite PRx’s promise, there are important limitations. So far, the literature is largely from observational studies. Therefore, it is unclear whether PRx defined autoregulatory failure is modifiable. 42 Autoregulatory failure could be triggered by the acute injury and result in profound physiologic derangements that may be irreversible. 43 Fortunately, work by Zeiler et al. suggests otherwise. For example, pharmacologic sedation may affect the amount of time patients spend with a PRx > 0. 44 While insufficiently studied, it seems intuitive that manipulating sedation could alter autoregulation by reducing cerebral metabolic rate and in turn, CBV.
Another limitation of PRx is related to its acquisition. Determining CPPopt comes from understanding how PRx changes across a CPP range and typically requires 4–6 hours of data sampling. This limitation means real time MAP adjustment may not be feasible as reported values reflect autoregulation from the previous 4–6 hours. To put it more simply, the moment to intervene may have already passed 4–6 hours ago during data acquisition. Another criticism is PRx curves require experts to interpret the data, and even then, there can be interobserver variability. 39 However, the use of the aforementioned multi-windowing method and computer identified PRx curves may mitigate these shortcomings.37,39 Finally, no RCTs have been conducted to provide an evidence based CPP guided algorithm clearly improves outcomes. As such, feasibility studies such as COGiTATE are underway. 45 In the meantime, PRx offers promise.
Cerebral microdialysis
Cerebral microdialysis (CMD) monitors cerebral metabolism during acute brain injury. Conceived in the 1970s by Ungerstedt & Pycock, CMD entered clinical use in 1989. 46 CMD uses a semi-permeable catheter, which is often combined with other cerebral monitors such as PbTO2, and ICP. Placed in the brain parenchyma, perfusate is instilled through a 10 mm catheter at a rate of 0.3 uL/min. This facilitates hourly sampling of small water-soluble molecules like lactate, pyruvate, glucose, glutamate or glycerol, as well as cytokines and larger proteins with the appropriate catheter.47,48
Molecules equilibrate with the microdialysate fluid and reflect the environment of the brain tissue surrounding the catheter. Microdialysate compounds are typically 20–100 kDa but equilibration depends upon molecule solubility, membrane area, membrane pore size, perfusate flow rate and perfusate choice. At a rate of 0.3 uL/min, the measured molecular concentrates are approximately 70%-reflective of the extracellular space. 49 By reducing the dialysate flow rate, recovery can be increased to 100%, but at the cost of sampling frequency. 50 In this article, we will focus on the commonly studied molecules such as lactate, pyruvate and glucose.
Glucose is the primary energy substrate of the brain. During aerobic metabolism, glucose is taken up by neural cells and metabolized to pyruvate, which, in turn, fuels the Kreb’s cycle.49,51 Following brain injury insult, neural cells may shift towards anaerobic metabolism, whereby, pyruvate is converted to lactate, which can, in turn, be measured at the bedside using CMD. In isolation, absolute lactate concentration is less useful than the lactate/pyruvate (L/P) ratio, as lactate may serve as an alternative neuronal energy source. However, a more detailed discussion of the lactate shuttle hypothesis is beyond the scope of this article.52,53 A widening L/P ratio (LPR) may signal an on-going cerebral energy crisis(i.e. ischemia or hypermetabolism) or physiologic distress.49,51,54 While it is unclear that adjusting physiologic variables to treat abnormal LPR values is consistently effective, CMD can be used in concert with PbTO2, and ICP monitors to potentially provide an understanding of the relationship between cerebral metabolism, cerebral oxygenation, ICP, and cerebral autoregulation.54,55
Neuroglycopenia, defined by a glucose of <0.8 mmol/L, may be caused by reduced glucose supply (i.e. from low cerebral blood flow, or low serum glucose), or increased demand (i.e. secondary to seizures, hyperthermia or cerebral hypermetabolism).51,56 During TBI, the normal cerebral glycemic supply-demand relationships are disturbed. Accordingly, neuroglycopenia has been observed in approximately 75% of patients with moderate-severe TBI at least once, 57 to which ICP and CPP monitoring were insensitive.57,58
Previously, clinicians strived for tight serum glucose control which was found to be harmful. 59 In TBI, CMD studies highlight that, when insufficiently monitored, tight glycemic control can also exacerbate cerebral metabolic crisis.60,61 This is clinically important because metabolic crisis, typically defined by a LPR >25–40 mmol/L, has been associated with increased mortality and worse neurologic outcomes at 3–6 months.54,62–67 The duration of metabolic crisis is also associated with an unfavorable neurologic outcome (GOSE < 6) at six months. 57 Even with a normal serum glucose, insulin infusions can substantially deplete cerebral glucose and potentially contribute to secondary injury. 68 Neuroglycopenia, even in the absence of cellular distress, is associated with poor neurologic outcomes at six months.61,69–73 Vespa et al. found in over 70% of cases of neuroglycopenia detected by CMD, there was no other physiologic indicator present(i.e. no systemic hypoglycemia, high ICP, low CPP, PbTO2, low jugular venous oxygen saturation, or seizure). 69 In contrast to neuroglycopenia, systemic hyperglycemia is also associated with poor outcome. 74 Therefore, similar to CPP, ICP and PbTO2, CMD could, hypothetically, be used to individualize serum glucose control to mitigate cerebral hypo- or hyperglycemia.
CMD has been studied in TBI and SAH to guide glucose control, CPP, hyperventilation, and temperature control.61,73,75–78 It has also been applied, albeit less commonly, following ischemic stroke, intracranial hemorrhage, hepatic encephalopathy, and epilepsy.78–82 Regardless, it is important to recognize the evidence for the use of CMD as a therapeutic monitoring tool remains relatively unsubstantiated. At the time of this writing, ‘Neuro-glycemic based’ insulin protocols should not be advocated for outside of the research context. Regarding their safety profile, CMD catheters appear to have a comparable complication risk to parenchymal monitors: approximately 1–3%.78,83 It would be rare for CMD to be used in isolation, and they are typically used in concert with PbTO2 and ICP monitors, which may be more sensitive compared to ICP monitors alone. 5
Outside of cerebral glycemic monitoring, CMD may help monitor the effects of temperature on cerebral metabolism. Oddo et al. showed improvements in cerebral metabolism following induced normothermia guided by CMD, but, notably, these gains were lost when shivering occurred.75,76 It remains unclear whether CMD guided cerebral temperature treatment changes outcomes, however the dangers of cerebral hyperthermia are well recognized.84,85 CMD could also be useful in predicting ICP spikes as the LPR >25 may predict intracranial hypertension. Accordingly, CMD monitoring could spur proactive, rather than reactive ICP management. 86 CMD could also help understand the role of brain cytokines, identify biomarkers, and help monitor intracerebral medication levels.87–91 Ultimately, the use of CMD remains largely investigative, however it does provide the potential to understand cerebral metabolism, which confers different neurophysiologic data from the other aforementioned techniques.
Conclusion
MMM, specifically PbTO2, PRx and CMD, are a promising group of techniques for understanding the complex neurophysiology which occurs during severe TBI. PbTO2 and PRx have accumulated enough evidence that experts advocate for goal directed therapeutic protocols targeting PbTO2 > 20 mmHg and CPPopt as part of clinical trials, while CMD remains largely investigational. Similarly, international guidelines from the BTF reflect these sentiments, however remain less rigorously defined to reflect the unclear state of the literature base as a whole.
In isolation, no monitoring tool is likely to change outcomes, but when used as part of a goal-directed therapeutic strategy, it is hypothetically possible to influence outcomes. 92 However, achieving this level of evidence starts from an applied physiologic question and sufficient supportive cohort studies, prior to the orchestration of RCT pilot studies and finally large therapeutic trials. In the case of MMM, BOOST-2 was successful in demonstrating protocolized PbTO2 & ICP treatment was feasible, safe and reduced the duration of cerebral hypoxemia. Accordingly, RCTs including BOOST-III, BONANZA and COGiTATE seek to substantiate the use of a protocolized treatment algorithms for PbTO2 & ICP directed care and CPPopt respectively. In the author’s opinion, supportive randomized trials are of great importance. These studies seek to demonstrate, not only, the proof of concept, but also to recognize unintended complications secondary to the therapies used (e.g. vasopressors, IV fluids, hyperoxia, etc).
Critical care practitioners should understand these tools should be used in an integrative fashion, combining MMM data with the clinical examination, systemic monitoring, neuroimaging, and additional specialized monitoring tools, such as EEG, transcranial doppler, etc., to enhance and individualize patient care. Clinicians do not merely want more data; we want information with which to monitor and individualize therapy meaningfully. MMM has not yet been shown to improve patient outcomes definitively. We need randomized trial data to show how to optimally guide use of MMM and enable better informed, individualized decisions at the bedside. However, as should be clear from this review, there is enough potential that this topic warrants ongoing attention. While we wait for randomized trial data, a case can be made for greater bedside brain monitoring and guarded clinical optimism.
Footnotes
Authors’ contributions
Acknowledgements: The authors would like to thank the Departments of Critical Care & Neurology at the University of Calgary, University of Alberta & Harvard University. Moreover, we would like to thank the reviewers and editorial team at Journal of the Intensive Care Society for their hard work and consideration.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.