
Abstract
While the single ant colony algorithm and the fish swarm algorithm have many advantages, they also have various shortcomings. After analyzing the advantages and disadvantages of the ant colony algorithm and the fish swarm algorithm, this paper uses the complementary principle of the two algorithms to effectively fuse the two population intelligent algorithms. The improved swarm intelligence algorithm is applied to the well-considered protein folding prediction problem, and the simplified protein structure Toy model is verified, and the ideal results are obtained. The improved algorithm enhances the search ability, and the computational efficiency is greatly improved, ensuring the accuracy of the operation.
Keywords
Introduction
Protein folding prediction in bioengineering has always been a focus of attention. According to the statistics of relevant departments, about one-third of the proteins in normal human cells will produce folding errors, but the intracellular mechanism will promptly discover the existence of these error proteins and quickly remove them. When the amount of protein folding is too large to be treated by these mechanisms, it can cause various diseases, similar to Alzheimer’s disease (commonly known as Alzheimer’s disease), cavernous hemangioma, family genetic cholesterol, rabies, etc. 1 Therefore, the study of protein folding is not only significant but also urgent.
Ant colony algorithm and artificial fish swarm algorithm (AFSA) are called two emerging cluster intelligence algorithms. Although it has not been long since they were born, the speed of their progress is very fast, and they have been tried by many scholars in the structural model of protein, and has achieved certain results.2–4 This paper analyzes the characteristics of the two algorithms and uses the principle of complementing the advantages and disadvantages to effectively fuse the two algorithms. This proves that this swarm intelligence performs better than a single algorithm and can achieve the optimal effect of the target problem.
Description of protein folding prediction
Protein folding structure prediction refers to predicting the natural structure of a protein from a given protein sequence. Proteins with a complete primary structure must have normal biological functions and must be folded into specific spatial structures. In other words, the biological properties of a protein are largely determined by the multidimensional spatial structure of its peptide chain. In the 1930s, Chinese scientist Wu Xian first pointed out that changes in the external environment can lead to the destruction of protein spatial structure and the loss of biological activity, but it does not destroy its primary structure. 5 The denatured protein usually becomes a stretched peptide chain, but under certain conditions, it can be refolded into the original spatial structure and restored to its original activity. This shows that the internal structural properties of the protein are contained in the protein chain, and the spatial configuration of the protein can be inferred through the protein chain. As early as 1973, Anfinsen put forward the famous theory that “the natural conformation of protein is the lowest conformation of free energy” in the journal “Science”, which laid the theoretical foundation for the prediction of protein folding structure, that is, by calculating the lowest energy of protein. Theoretical basis for predicting protein thermodynamics structure. 6 Therefore, from a mathematical point of view, we can attribute the problem of protein folding structure prediction to a global optimization problem.
The general flow of protein structure prediction is to first analyze the sequence of a protein after a given protein sequence, and find a homologous protein in a library of known protein sequences. If found, and the spatial structure of the protein is determined, it is possible to use homologous information to establish a tertiary structure model of the target protein. However, in actual research, we generally first predict the secondary structure of the protein, and then predict the tertiary structure. The accuracy of the tertiary structure prediction depends on the results of the secondary structure prediction, see in Figure 1.5 Searching the entire conformational space of the protein finds the lowest energy conformation as the conformation of the target protein, which is the protein folding prediction.
At present, the main problems in protein structure prediction are: the accuracy of prediction is not high enough, the calculation speed is not fast enough, and there is a certain distance between the model of structure prediction and real protein. In recent years, the swarm intelligence has brought new hopes for solving the above problems, especially in the aspect of solving complex problems, which shows the characteristics of dynamic, adaptability, and robustness, which helps to compensate for the existing protein structure prediction problems, weaknesses and defects. In recent years, with the rise of various intelligent random algorithms and widely used in various fields, it has been found that the use of these algorithms to solve the problem of protein folding has unique advantages. In this paper, a new algorithm combining ant colony and AFSA is applied to the protein folding prediction problem, and good results have been achieved.
Principles of ant colony algorithm
In many years of research, biologists have discovered that ants in nature are invisible, and that the intelligence and mobility of a single ant are negligible. Only relying on communication with peers, as well as the special material left behind when walking—pheromone to change their direction of action. 7 Based on this, Italy’s Dorgio and its research team proposed an algorithm in the early 1990s, named Ant Colony Optimization (ACO). 8 The algorithm is also called ant colony optimization and is a probability algorithm. This is a general heuristic method. The concept of ant colony algorithm is convenient to understand, it has a few of parameters and does not involve complex situations such as mutation and crossover. 9 The application field of the algorithm is very wide. From appearance to present, research on ant colony algorithms continues to rise in various industries, and the algorithm can be seen in almost every optimization field. 10
Description of ant colony algorithm
When the ant passed from

In which,

In formula (2),

In the formula, when the ant
Thoughts on improving ACO–AFSA algorithm
The ant colony algorithm has good parallelism and high precision of the algorithm, which is a prominent advantage of the ant colony algorithm. Artificial ants can search the entire search space, and information exchange between artificial ants can be achieved, and finally the purpose of optimization is achieved. In the ant colony algorithm, in fact, each artificial ant interacts with the environment, and each artificial ant is connected to each other by means of pheromone “bridge”. Specifically: ants must first have some kind of inertia, keep moving forward, rather than walking around. Second, after the ants have fixed the trend of movement, they must also maintain a certain degree of randomness. Taboo, like the particles in the particle swarm algorithm, has been moving in a crazy linear motion. 11 This allows the ants to do their best to follow the original trend and try to generate new search ranges. But ants also have mistakes, so do not move to places with high pheromones, and walk blindly, which produces a local optimal situation, which is also a shortcoming in the ant colony algorithm.
The AFSA is a group intelligent bionic algorithm that simulates the behavior of fish in nature. By optimizing the four underwater behaviors of a single artificial fish, local optimization is achieved, and finally the global optimum is achieved. In the fish’s eating behavior, the number of trials is set less, the probability of artificial fish random walk is increased, the ability of local optimal solution can be avoided, and the global optimality is obtained.12,13 The use of the crowding factor can limit the size of the fish population, which allows the artificial fish to search more freely. The occurrence of trailing behavior can make the artificial fish move toward a better state, and can also cause the artificial fish caught in the local optimum to escape from the local optimal region and move toward the global optimal direction. A typical disadvantage of the AFSA is its low accuracy.
Through the above description, it can be concluded that the ant colony algorithm and the AFSA have complementary relationships, and the intelligent algorithms of the two populations are improved, and the effective fusion can improve the performance and efficiency of the algorithm. The improved group intelligence algorithm can avoid the disadvantages of the single algorithm, strengthen the advantages of the algorithm, and form a more stable and comprehensive group intelligent algorithm.14
Steps to improve the ACO–AFSA algorithm
This paper is based on a single ant colony algorithm and AFSA to improve and merge into a new algorithm—ant colony fish swarm algorithm, referred to as ACO–AFSA. The steps of the ACO–AFSA algorithm are as follows:
Step 1: Initial setting of the ant colony algorithm; Step 2: Initial setting of the AFSA; Step 3: Generate a new initial solution; Step 4: Calculate fitness; Step 5: Update pheromone concentration and bulletin board information; Step 6: Determine if the condition is met.
Now, we will elaborate on the above steps.
Initialize the number of artificial ants m, heuristic factor, hope heuristic factor, information persistence factor, and information intensity Q. Initially set the number of artificial fish M, step, artificial fish field of view visual, congestion factor. The ant colony algorithm and the AFSA alternately iterate, and use comparison to determine the superiority of the solution. In the same problem, if the ant colony algorithm first searches for a good solution, it is randomly placed in an artificial fish position in the artificial fish group. If the AFSA first searches for a good solution, then it is randomly placed in an artificial ant position in the artificial ant colony. Meanwhile, judge whether it is crowded according to the formula Get the adaptation value according to the formula Follow the instructions to update the pheromone concentration and update the bulletin board information. Determine whether the conditions for ending the algorithm process are met. If it is satisfied, the output solution is the optimal solution. If it is not satisfied, return to step 3 to continue the operation.
Prediction of protein folding in the Toy model
Protein folding prediction faces two major problems. One is how to convert the cumbersome protein structure into a simple mathematical model to simplify the problem; the other is how to find efficient search methods to efficiently search for protein conformations. 16 At present, the simplified protein structure models mainly include Toy model, H-hydrophobic amino acid, P-hydrophilic amino acid (HP) grid model, three-dimensional cubic model, and hexagonal lattice model. 17 In this paper, the improved ant colony fish swarm algorithm is applied to the protein folding prediction problem of the two-dimensional Toy model, and the effect is ideal.
Two-dimensional Toy model
The Toy model was proposed by Stillinger and his companions in the 1990s and is now also known as the AB non-grid model. 18 In this model, the common 20 amino acids are represented by B (hydrophilic or non-polar) and A (hydrophilic or polar) to represent hydrophilic amino acids and hydrophobic amino acids. 19 The adjacent amino acids are connected by a steel bond, and the angle between the two amino acid atoms can be formed, that is, the bond angle, as shown in Figure 2.

General process for protein structure prediction.

A Toy model for protein folding.
The amino acid sequence in Figure 2 is nine and has seven angles, respectively
No matter how long the amino acid sequence is, their energy functions are the same, which is the sum of Van der Waals gravitational potential energy and bending potential energy.
20
The length of a protein is N, and the Van der Waals potential between the separated residues is

These three values represent strong attraction between AA, mutual exclusion between AB, and weak attraction between BB, showing the nature of the protein from the side. 21 It can be seen from equation (4) that Van der Waals gravitational potential energy is determined by the hydrophobicity, polarity, and distance of non-adjacent residues.
The main chain bending potential energy is

It can be seen from equation (5) that the magnitude of the potential energy and the protein sequence itself are not determined by the chain

In the two-dimensional Toy model, this paper transforms the protein folding prediction problem into a function optimal value problem, that is, how to obtain N-2 bond angles to minimize the energy value. 22 In this way, we abstract the abstraction into concrete, simplify the complex problem, and change the structure of the real protein without changing its characteristics.
Prediction of two-dimensional Toy models protein folding
When using the Toy model for protein folding prediction, the most common method is to use the Fibonacci sequence as the amino acid sequence of the protein. The Fibonacci sequence is also called the golden section, and the form of the series is: 0, 1, 1, 2, 3, 5, 8, 13, 21… .
23
The third item of the series begins with each item being the sum of the previous two items, which is expressed as equation (7)

When performing protein folding prediction, convert it to
Table 1 shows the lengths of the protein sequences calculated using the Fibonacci sequence, which are 5, 8, and 13, respectively. This paper experiments on Intel (R) Core i3 2.40 GHz CPU, 2.00GB RAM, and 32-bit operating system, using MATLAB7.0 software to test these three sequences. The important parameters in the algorithm are set to:
Fibonacci series.

Test result.

The protein folding prediction of the two-dimensional Toy model was performed using the improved algorithm of this paper. Under the premise of guaranteeing the quality of the solution, the three sequences can find the lowest energy value. In order to increase the persuasiveness of the improved ant colony fish swarm algorithm in this paper, it is indicated that the improved algorithm can be applied to the protein folding prediction problem. The results of other literature calculations are listed below, as shown in Table 3.
Result comparison.

Source: reproduced with permission from Hou, 2014. 24
It can be seen from Table 3 that with the improved algorithm of this paper, the energy value can also be obtained, and the effect is higher than that of the existing research. In order to explain more intuitively the results of this paper, the experimental results will be compared with the line graph, as shown in Figure 3. It can be seen from the line graph of Figure 3 that the new algorithm has a lower trend than the original algorithm. It shows that the convergence speed of the algorithm is faster, and new algorithm can find better results in a shorter time. The structure of real proteins is extremely simple, using computer simulations of protein structures for protein folding prediction. Although the true structure of the protein cannot be completely reflected, the energy value can provide a reliable reference value for the stability of the natural protein structure. The data shown in the table not only show the feasibility of the algorithm, but also show its effectiveness. The energy constellations of these three sequences are shown in Figures 4 to 6. Through these three energy constellation diagrams, we can also find that the longer the energy constellation of the sequence, the better the effect, and the closer it is to the appearance of the real protein structure.

Comparison of experimental results.

A constellation of length 5.

A constellation of length 8.
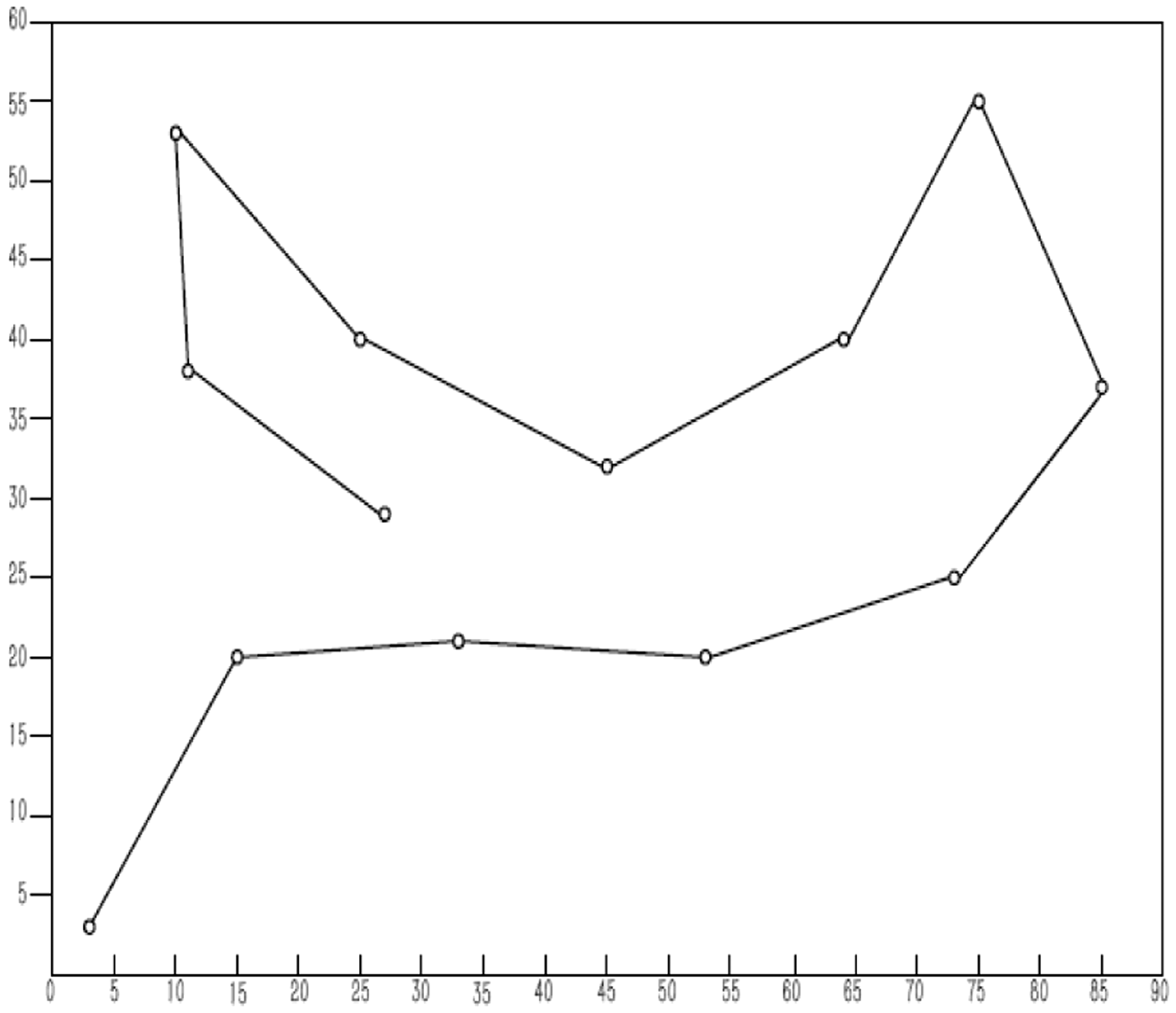
A constellation of length 13.
Applying the improved ant colony fish swarm algorithm to the two-dimensional Toy model in the protein folding prediction problem, the Fibonacci sequence is tested and the results are valid. Compare the test results with the results in other literatures, and draw a constellation diagram, the effect is outstanding. The results of this paper are slightly better, and the conformational map can basically reflect the physical properties of proteins.
Conclusion
Protein folding prediction is a very hot topic in the field of bioengineering, involving many aspects. The use of swarm intelligence algorithms for protein folding prediction is a novel topic and a trend for future research. In this paper, the improved ant colony fish swarm algorithm is applied to the protein folding prediction problem. The artificial protein Fibonacci sequence was tested using a two-dimensional Toy model. The test results are compared with the results in other literature and an energy conformation map is given. There are still many places in this paper that can be improved through research. The improved algorithm in this paper can also be applied to higher domain layers through deformation. These ideas will be verified in later research.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The paper is supported by Heilongjiang University education reform project (SJGY20180565), 2018 Annual Education Department Basic Research Business Youth Innovation Talent Project (135309376), Intelligent Manufacturing Equipment Innovation Team--Heilongjiang Province Intelligent Manufacturing Equipment Industrialization Collaborative Innovation Center (135409102), and 2019 Qiqihar City-level Science and Technology Plan General Project Contract Number (GYGG-201919).