
Abstract
An oxothiazolidinylidene derivative, Ethyl (Z)-2-((Z)-3-(4-acetamidophenyl)-2-(((E)-1-(4-acetamidophenyl)ethylidene)hydrazono)-4-oxothiazolidin-5-ylidene)acetate (

Keywords
Introduction
Although epidermal growth factor receptor (EGFR) is a validated therapeutic target,1,2 resistance mutations – particularly T790M – limit the long-term efficacy of first-generation tyrosine kinase inhibitors (TKIs). Therefore, the development of mutation-tolerant inhibitors remains a significant challenge.3,4
Computational drug design has become an essential component of modern drug discovery, integrating structure-based design, molecular docking, density functional theory (DFT), and molecular dynamics (MD) simulations to optimize structural and electronic properties.5–8
In our recent work, computational approaches were employed to rationalize the design strategy in the context of EGFR inhibition. DFT calculations were utilized to evaluate electronic reactivity and molecular stability, while molecular docking was applied to assess compatibility within the ATP-binding pocket. MD simulations were conducted to examine conformational stability and potential mutation tolerance, particularly in resistant EGFR variants. In addition, Molecular Mechanics/Generalized Born Surface Area (MM-GBSA) analysis provided quantitative estimation of binding energetics to support structure–activity interpretation.9–13 When paired with in silico ADME-Tox evaluations, these methods allow the identification of promising drug-like candidates while reducing experimental time, cost, and effort.14,15
In this context, oxothiazolidinylidene derivatives have attracted growing attention due to their versatile biological activities, including antitumor, 16 anti-inflammatory, 17 and antimicrobial effects.18–20 The oxothiazolidinylidene core, when linked with heteroaromatic or hydrazono moieties, has been shown to enhance molecular flexibility and facilitate key hydrogen bonding and π–π stacking interactions within the ATP-binding cleft of kinases, thereby improving inhibitory potency and mutation tolerance.21,22 Although oxothiazolidinylidene derivatives had previously been explored for various biological activities, their application as EGFR-targeting inhibitors remained limited. Most reported EGFR inhibitors (EGFRIs) relied on classical hinge-binding heterocycles and established pharmacophoric arrangements. In contrast, Z-EAHT introduced a hybrid oxothiazolidinylidene framework that was strategically designed to maintain hinge-region anchoring while extending hydrophobic interactions within the ATP-binding pocket.
This structural configuration represented a deviation from conventional EGFRI scaffolds and offered a distinct interaction profile that may have contributed to improved binding stability.
Rationale
EGFRIs share four key pharmacophoric features essential for binding to the EGFR active site. Using Erlotinib I as an example: (1) a planar heteroaromatic system that fits into the adenine-binding pocket for π–π stacking, (2) a terminal hydrophobic head occupying hydrophobic region I to enhance affinity, (3) a linker NH group forming hydrogen bonds in the linker region, and (4) a hydrophobic tail extending into hydrophobic region II for complex stabilization23–27 (Figure 1(a)).

(a) Core pharmacophoric characteristics of known EGFR inhibitors. (b) Strategy for developing a
Our group previously developed a thiadiazole-based EGFRI (N-ASTPC) that exhibited encouraging activity, motivating further structural refinement toward improved mutation-tolerant EGFR inhibition.
10
Based on its promising results,
To the best of our knowledge, this study represented an oxothiazolidinylidene-based scaffold demonstrating comparable sub-nanomolar inhibitory potency against both wild-type EGFR (EGFR-WT) and the resistant EGFR-T790M mutation. Unlike previously reported EGFR inhibitors, this work integrated long-timescale MD simulations, free energy decomposition, ProLIF interaction profiling, principal component analysis of trajectories (PCA-T), and free energy landscape (FEL) analyses with experimental validation, providing comprehensive mechanistic insight and establishing Z-EAHT as a mutation-tolerant EGFR inhibitor candidate.
Results and discussion
In silico investigations
DFT calculations
DFT were conducted using the B3LYP/6-31 G + (d, p) level of theory within the Gaussian 09 software. The molecular structure of

Optimized structure (a), HOMO and LUMO distributions (b), the plot of TDOS (c), Mulliken charge distribution (d), and ESP (e) at the B3LYP/6-31G+ (d, p) level for
The analysis of frontier molecular orbitals (FMO) sheds light on the electronic configuration and reactivity of molecules, which is essential for comprehending their interactions with biological targets. FMO analysis examines the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) to reveal reactive regions. The energy gap between these orbitals indicates chemical stability, with smaller gaps suggesting higher reactivity. FMO analysis helps predict molecular interactions by identifying nucleophilic or electrophilic sites, aiding drug design in biochemical systems. 28
This is depicted in Figure 2(b), the HOMO is predominantly located on the electron-rich nitrogens of hydrazine, nearby aromatic rings, and the acetamidophenyl group, suggesting that these areas serve as electron donors when interacting with the target protein. The LUMO is distributed over the thiazolidinone moiety and carbonyl, indicating their roles as electron acceptors. This specific HOMO and LUMO distribution facilitates charge transfer throughout the molecular structure and predicts significant chemical interactions with the biological target. The HOMO/LUMO gap is shown in Figure 2(b) was calculated to be relatively small (3.318 eV), indicating efficient electron mobility across
Table 1 presents the calculated global chemical parameters for
The DFT calculated global reactivity parameters for

As shown in Figure 2(d), the electron density is distributed throughout
The mapping of the Electrostatic potential (ESP) surface of
Molecular docking
To investigate the potential mechanism of EGFR inhibition by

(a) 3D and 2D interactions of
Comparative docking analysis of

MD simulations
MD simulations demonstrated notable conformational stability and distinct dynamic behavior of the EGFR protein upon complexation with
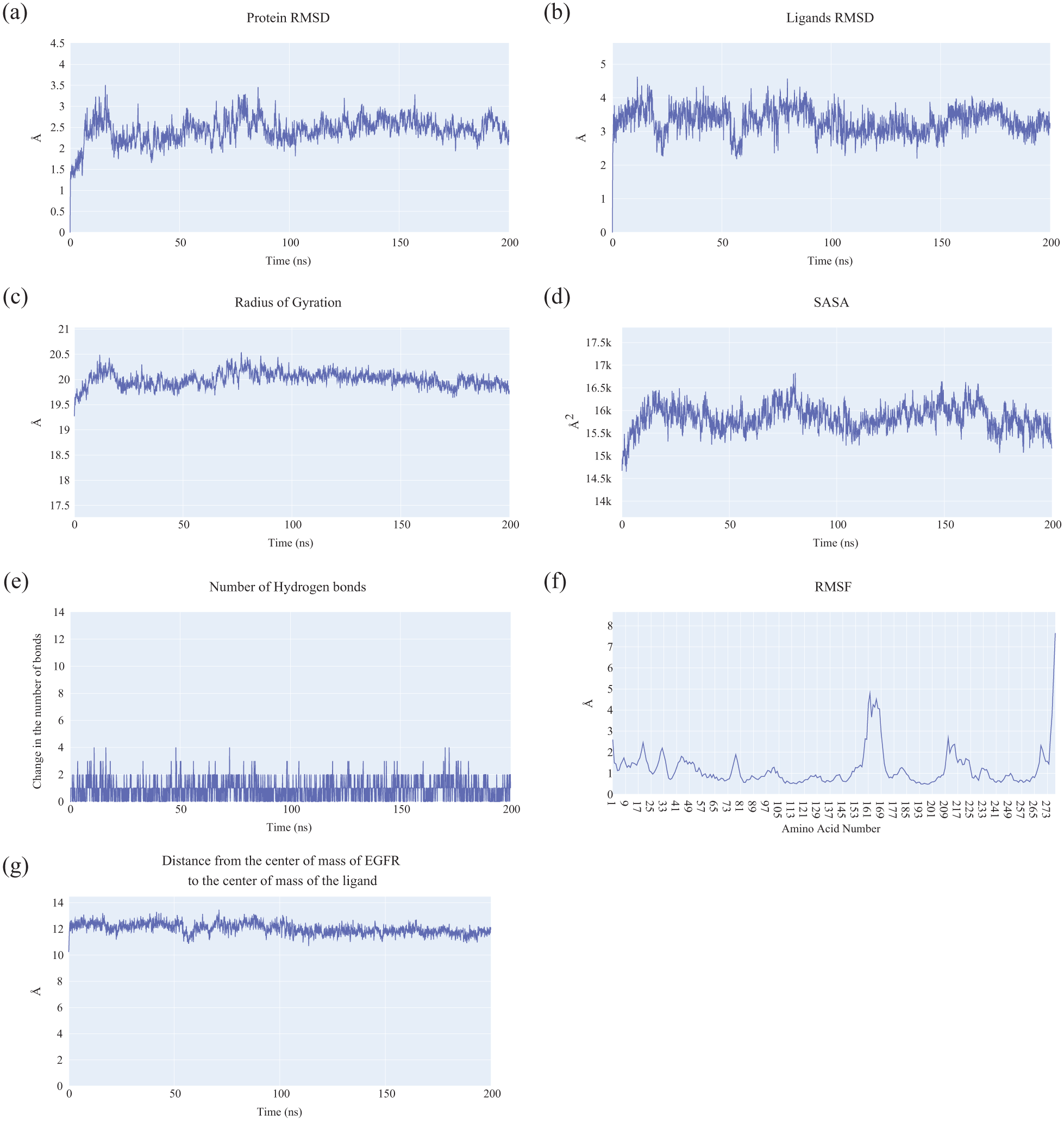
Molecular dynamics analyses of the EGFR–Z-EAHT complex: (a) RMSD profile of the EGFR backbone, (b) RMSD of Z-EAHT, (c) RoG of EGFR, (d) SASA of EGFR, (e) fluctuations in the number of hydrogen bonds, (f) RMSF of EGFR residues, and (g) center-of-mass distance between Z-EAHT and EGFR.
Further structural evaluations using the radius of gyration (ROG) (Figure 4(c)) and solvent-accessible surface area (SASA) (Figure 4(d)) indicated no substantial alterations in the protein’s compactness or solvent exposure, implying that ligand binding did not induce major conformational rearrangements in EGFR. Hydrogen bond analysis (Figure 4(e)) revealed the presence of an average of two persistent hydrogen bonds between
Residue-level flexibility, as depicted by the root mean square fluctuation (RMSF) of C-alpha atoms (Figure 4(f)), showed minimal deviations ranging from 1 to 1.8 Å across most residues, reflecting overall rigidity of the protein-ligand complex. Notably, residue ALA847:HB2 exhibited a higher RMSF value of 8.29 Å, possibly indicating localized flexibility or transient conformational movement in that region. The center-of-mass distance analysis (Figure 4(g)) confirmed consistent ligand positioning, maintaining a stable average separation of approximately 12 Å until the end of the simulation.
Collectively, these results confirm that the EGFR_
Figure 5 summarized the MM-GBSA binding free energy analysis of the EGFR–Z-EAHT complex. The calculated total binding free energy was −40.01 kcal/mol, indicating a strong and favorable interaction. Van der Waals (−54.9 kcal/mol) and electrostatic contributions (−29.8 kcal/mol) were identified as the primary stabilizing forces, highlighting the synergistic role of nonpolar and polar interactions within the EGFR binding pocket (Figure 5(a)).
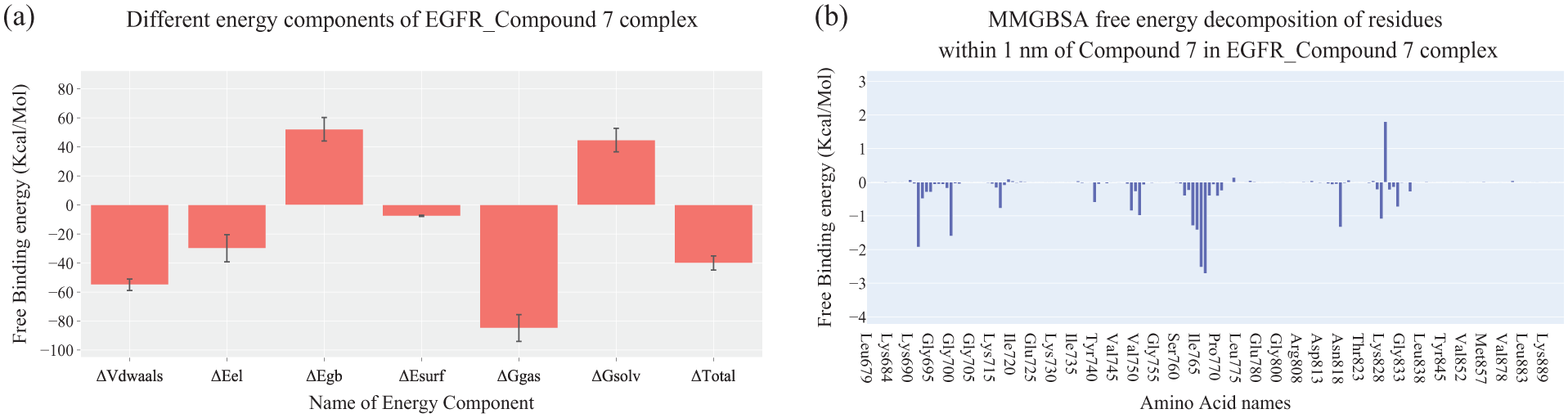
(a) Individual MM-GBSA energy components and their corresponding values for the EGFR–Z-EAHT complex. (b) Per-residue binding free energy analysis of the EGFR–Z-EAHT complex.
Per-residue energy decomposition analysis of amino acids located within 1 nm of the ligand further identified key contributors to binding stabilization (Figure 5(b)). Notably, Thr766, Gln767, Lys692, Ile765, Leu764, Asn818, and Gly700 exhibited favorable energetic contributions, while ILE829 showed a minor unfavorable contribution, possibly due to steric or electrostatic effects.
Overall, the MM-GBSA results confirmed that Z-EAHT achieved stable binding within the EGFR active site, predominantly driven by van der Waals and electrostatic interactions.
Protein–ligand interaction fingerprint (ProLIF) analysis provided additional insight into the residue-level interactions governing Z-EAHT recognition within the EGFR binding pocket. As presented in Supplementary Data (Fig. S2.2.1 and S2.2.2), several residues exhibited high-frequency hydrophobic contacts (>90%) throughout the simulation, including Leu694, Val702, Ala719, Cys791, and Met769. Notably, Met769 also demonstrated significant hydrogen bond acceptor and van der Waals interactions, indicating its key stabilizing role. These findings indicate that persistent hydrophobic contacts substantially contributed to complex stabilization, complementing the energetic profile obtained from the MM-GBSA analysis.
PCA-T was performed to investigate the dominant collective motions of the EGFR–
Projection of the trajectory onto the PC1–PC2 plane revealed well-defined and recurrent low-energy basins (Figure 6), indicating stable conformational sampling throughout the simulation. The presence of shallow energy barriers between adjacent minima suggested smooth transitions between closely related metastable states without structural destabilization. For completeness, additional projections onto the PC1–PC3 and PC2–PC3 planes are provided in Supplementary Data (Fig. S2.2.4).

Two- and three-dimensional FEL projections of the EGFR–
Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profiling study
The comparative ADMET analysis between
Predicted ADMET properties of

In silico toxicity studies
The predicted toxicity parameters of
In silico toxicity and safety profile of

mg/kg/day.
g/kg.
Furthermore, the rat oral LD₅₀ of
Overall, the toxicity assessment supports
Synthesis
The design and synthesis of the EGFR (epidermal growth factor receptor) enzyme inhibitor were achieved through a systematic synthetic route. Initially, 4-aminoacetophenone

The structural integrity of the synthesized compound was confirmed through comprehensive spectroscopic and analytical characterization. The FTIR spectrum displayed characteristic absorption bands corresponding to NH stretching (3328 and 3204 cm-1), carbonyl (C=O) stretching (1684 cm-1), and imine (C=N) functionality (1593 cm-1), consistent with the proposed structure. The 1H NMR spectrum showed two distinct singlets attributable to NH protons along with well-resolved aromatic and aliphatic signals matching the expected proton environments. The 13C NMR spectrum further confirmed the presence of carbonyl, aromatic, and aliphatic carbons at the anticipated chemical shifts. Elemental analysis values were in close agreement with the calculated composition, confirming molecular integrity. In addition, high-performance liquid chromatography (HPLC) analysis indicated 100% purity, demonstrating the absence of detectable impurities.
Biological evaluation
Enzyme inhibition
The EGFR inhibitory potency of the newly designed compound
EGFR inhibitory activity (IC₅₀, µM) of

The enhanced potency of
Overall, these results indicate that
Cytotoxicity
The in vitro cytotoxicity evaluation of
Cytotoxic activity (IC₅₀, µM) of

In contrast,
Collectively, these findings confirm that
Immunofluorescence analysis
The immunofluorescence analysis (Figure 7) illustrates the effect of
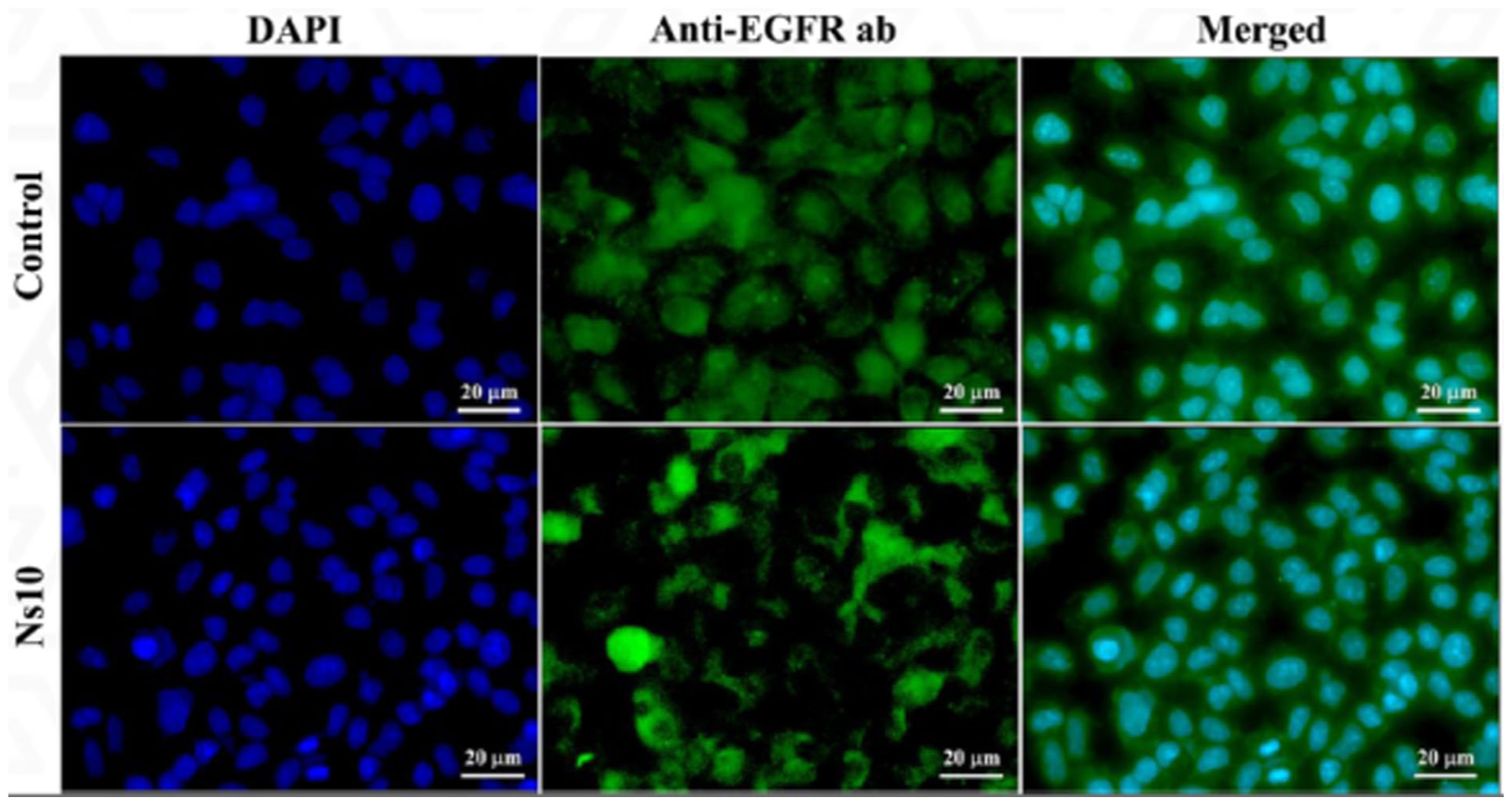
Immunofluorescence analysis showing the effect of
Quantitative analysis further supports these observations: EGFR expression in the
Quantification of apoptosis-related proteins using enzyme-linked immunosorbent assay
The apoptotic induction potential of
Effect of

Concentrations and fold-level differences (FLD) of Bax, Bcl-2, and Caspase-3 relative to untreated control.
Specifically, Bax expression increased from 50.77 ng/mL in control cells to 283.99 ng/mL following
Furthermore, Caspase-3, the major executioner caspase, showed a substantial increase from 53.29 to 308.9 ng/mL (5.80-fold elevation), supporting that
Taken together, these findings suggest that
Correlation between in silico and experimental findings
A comparison between the in silico predictions and the experimental findings revealed a strong correlation. Z-EAHT exhibited a favorable docking score (−20.99 kcal/mol) and stable binding interactions within the EGFR-WT active site, which is consistent with its potent in vitro inhibitory activity (IC50 = 0.012 µM). The preservation of the critical hinge-region hydrogen bond and extensive hydrophobic interactions may account for the observed biological potency. Furthermore, the predicted ADMET profile supported acceptable drug-like properties, aligning with the experimental performance of the compound. Overall, the computational findings successfully rationalize and support the experimental results.
Materials and methods
Chemistry
Chemicals, reagents, and instrumentation
All reagents and solvents employed in the synthesis of
Synthesis of Z-EAHT
A general procedure for the synthesis of intermediates 2 and 4
Intermediates
Synthesis of methyl hydrazinecarbodithioate 3
The reagent was prepared using a typical technique authenticated in literature.11,30–33
Synthesis of ethyl (Z)-2-((Z)-3-(4-acetamidophenyl)-2-(((E)-1-(4-acetamido phenyl) ethylidene)hydrazono)-4-oxothiazolidin-5-ylidene)acetate (Z-EAHT)
In a 100-mL oven‑dried round‑bottom flask equipped with a magnetic stir bar, the intermediate (0.01 mol) was dissolved in methanol (10 mL), followed by the addition of Diethyl acetylenedicarboxylate (DEAD, 0.01 mol, 1 mmol). The reaction mixture was stirred at room temperature for 8 h. Upon completion, the precipitated product was collected by filtration, purified through recrystallization from an appropriate solvent, and obtained as
Color: yellow powder; Yield: 78%; m.p.: 195-197°C, FTIR (KBr, νmax/cm⁻¹); 3328, 3204 (2NH), 3061, 3003 (CH-aromatic), 2789 (CH-aliphatic), 1684 (C=O), 1593 (C=N), and 1566 (C=C); HPLC purity 100%; 1H NMR (400 MHz, DMSO-d6) δH = 10.17 (s, 1H, NH(24)), 10.16 (s, 1H, NH(34)), 7.82 (d, J = 8.4 Hz, 2H, ArH(18, 20)), 7.70 (dd, J = 18.9, 8.4 Hz, 4H, ArH(17,19,27,29)), 7.43 (d, J = 8.5 Hz, 2H, ArH(26,30)), 6.77 (s, 1H, C=CH(2), 4.28 (q, J = 7.1 Hz, 2H, OCH3CH2(35)), 2.22 (s, 3H, CH3(14)), 2.08 (d, J = 5.9 Hz, 6H, 2CH3(22,32)), 1.29 (t, J = 7.1 Hz, 3H, OCH2CH3(36)); 13C NMR (101 MHz, DMSO) δC = 169.14 (C31), 166.01 (C21), 164.58 (C1), 164.14 (C8), 158.88 (C13), 142.31 (C6), 141.93 (ArC18), 140.09 (C9), 131.88 (ArC18), 129.25 (ArC15), 128.74 (ArC25), 127.85 (ArC16,20), 119.55 (ArC26,30), 119.02 (ArC27,29), 115.55 (C2), 61.91 (C35), 24.55 (C22), 24.51 (C32), 15.15 (C14), 14.50 (C36); Anal. calcd for C25H25N5O5S (507.57): C, 59.16; H, 4.96; N, 13.80. Found: C, 59.43; H, 5.12; N, 14.03%.
DFT calculations
The molecular geometry of
Molecular docking studies
Molecular docking of
MD simulations
The EGFR–
Binding free energy analysis (MM-GBSA)
Binding free energies were calculated using the MM-GBSA approach implemented in gmx_MMPBSA. A per-residue energy decomposition analysis was performed to identify amino acid residues within 1 nm of the ligand that significantly contribute to the stability of the EGFR–
ProLIF interaction analysis
Interaction profiling was performed using the ProLIF Python package to quantitatively and qualitatively assess ligand–residue interactions throughout the 200 ns simulation. Key interaction types—including hydrogen bonding, π–π stacking, π–cation contacts, hydrophobic interactions, and salt bridges—were identified, and their occupancy frequencies were quantified across all trajectory frames. 43 Complete analysis parameters are provided in Supplementary Information.
PCA-T
EGFR α-carbon mobility was analyzed through PCA-T based on the mass-weighted covariance matrix (C). Trajectories were aligned to the equilibrated reference structure of the EGFR–
FEL analysis
FEL mapping was performed to visualize the conformational states sampled during MD simulation. The landscape was projected onto two principal reaction coordinates, reflecting the dominant motions. The free energy (Gα) associated with each conformation was computed from the Boltzmann distribution, linking conformational probability to thermodynamic stability. 47 Detailed FEL plots are provided in Supplementary Information.
In Silico ADMET and toxicity prediction
ADMET and toxicity profiles were predicted using BIOVIA Discovery Studio. Parameters assessed included Ames mutagenicity, carcinogenicity, rat LD₅₀, rat LOAEL, maximum tolerated dose, DTP toxicity, and skin/ocular irritancy. Comparative ADMET data for
In vitro EGFR enzyme inhibition assay
EGFR inhibition assays for
In vitro cytotoxicity and selectivity assays
Cytotoxicity was assessed using the MTT assay against HepG-2, MCF-7, HCT-116, PC-3, and A549 cancer cell lines, along with the normal WI-38 fibroblast line. Cells were treated with increasing
Apoptosis-related protein quantification (enzyme-linked immunosorbent assay)
The levels of Bax, Bcl-2, and Caspase-3 proteins in A549 cells treated with
Conclusion
In this study, a novel oxothiazolidinylidene hybrid,
Supplemental Material
sj-pdf-1-chl-10.1177_17475198261442267 – Supplemental material for An EGFR-Targeting Oxothiazolidinylidene Derivative: From Computational Design to Experimental Validation
Supplemental material, sj-pdf-1-chl-10.1177_17475198261442267 for An EGFR-Targeting Oxothiazolidinylidene Derivative: From Computational Design to Experimental Validation by Eslam B Elkaeed, Hazem Elkady, Walid E Elgammal, Ahmed Nofal, Hazem A Mahdy, Dalal Z Husein, Fatma G Amin, Aisha A Alsfouk, Ibrahim H Eissa and Ahmed M Metwaly in Journal of Chemical Research
Footnotes
Ethical considerations
Not Applicable.
Consent to participate
Not Applicable.
Consent for publication
Not Applicable.
Author contributions
A.A.A. contributed to funding acquisition as well as manuscript writing and revision. H.A.M., H.E., W.E.E., and A.N. performed the chemical synthesis as well as contributed to manuscript writing and revision. E.B.E. contributed to manuscript writing and revision. F.G.A. carried out the MD simulations, while D.Z.H. conducted the DFT calculations. I.H.E. also contributed to the experimental methodology, ensuring the availability of essential synthesis materials. A.M.M. supervised the project, designed the study, and prepared the initial manuscript draft. All authors reviewed and approved the final manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2026R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are enclosed in the manuscript and the supplementary materials.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.