
Abstract
The co-expression of epidermal growth factor receptor and carbonic anhydrase-IX in aggressive tumors such as triple-negative breast cancer contributes to therapeutic resistance through proliferative signaling and hypoxia-mediated adaptation. Dual inhibition of these targets represents a promising strategy for overcoming tumor resilience and enhancing treatment efficacy. A novel thiadiazole-based hybrid molecule (N-(4-((E)-1-(((Z)-5-acetyl-3-(4-sulfamoylphenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)benzamide), ASTPB, was rationally designed through pharmacophore modeling, followed by molecular docking and molecular dynamics simulations for 200 ns to assess its dual-binding affinity to epidermal growth factor receptor and carbonic anhydrase-IX. Molecular dynamics simulations confirmed the stability of ASTPB complexes with both epidermal growth factor receptor and carbonic anhydrase-IX, as indicated by stable root mean square deviation values, consistent radius of gyration, and persistent hydrogen bonding, which supported its strong dual-binding affinity. Further quantum chemical analysis via density functional theory supported the electronic stability and reactive potential of ASTPB. In addition, in silico ADMET (Absorption, Distribution, Metabolism, Excretion, and toxicity) predictions indicated low toxicity, a wide safety margin, limited solubility and absorption, and overall favorable metabolic and distribution profiles. ASTPB was synthesized and exhibited potent dual inhibition of carbonic anhydrase-IX and epidermal growth factor receptor, with IC₅₀ values of 0.046 ± 0.007 µM and 0.059 ± 0.009 µM, respectively, comparable to standard inhibitors. In cytotoxicity assays, ASTPB was examined across several cancer cell lines (HePG-2, MCF-7, HCT-116, PC-3, MDA-MB-231), with the greatest efficacy against MDA-MB-231 (IC₅₀ = 18.15 µM) and minimal toxicity toward normal WI-38 cells (IC₅₀ = 72.46 µM). Cell cycle analysis revealed G0/G1 phase arrest, while Annexin V/PI staining indicated early apoptosis in 76.7% of cells. Molecular assays confirmed upregulation of Bax and caspase-3 (3.7-fold each) and downregulation of Bcl-2 (1.97-fold), indicating activation of the intrinsic apoptotic pathway. In conclusion, ASTPB is a promising dual carbonic anhydrase-IX/epidermal growth factor receptor inhibitor with selective cytotoxicity and potent pro-apoptotic activity, particularly in triple-negative breast cancer models. These findings support its potential as a multi-targeted therapeutic lead in the treatment of hypoxia-adapted, epidermal growth factor receptor–driven malignancies.

Introduction
Cancer remains a leading cause of mortality worldwide, driven in part by the complexity and adaptability of tumor biology. 1 One of the hallmarks of aggressive and treatment-resistant cancers is the co-expression of multiple oncogenic drivers, which contribute synergistically to tumor progression, metastasis, and therapy evasion.2,3 In particular, EGFR and CA-IX represent two clinically validated yet mechanistically distinct targets that are often co-upregulated in hypoxic, rapidly proliferating tumors, such as triple-negative breast cancer (TNBC),4–6 hepatocellular carcinoma,7–9 and certain colorectal malignancies.10–12
EGFR, a member of the ErbB receptor tyrosine kinase family, plays a pivotal role in controlling cell proliferation, survival, and migration through the activation of several downstream signaling cascades. 13 Aberrant EGFR activity is implicated in oncogenesis, tumor aggressiveness, and resistance to therapy. 14 On the other hand, CA-IX is a transmembrane metalloenzyme upregulated under hypoxic conditions.15,16 It facilitates extracellular acidification and intracellular pH homeostasis, promoting tumor cell survival, invasiveness, and immune evasion in acidic microenvironments.17–19
Combining in silico and in vitro methodologies provides a powerful and streamlined platform for modern drug discovery.20,21 Computational tools facilitate the prediction of molecular binding behaviors, interaction energies, and pharmacokinetic profiles, while biological assays validate these predictions and offer critical mechanistic insights.22,23 The synergistic use of these approaches not only expedites the screening and refinement of therapeutic candidates but also enhances the robustness of findings and minimizes reliance on time-intensive and costly in vivo experimentation. 22 Our research group has effectively applied this integrated strategy across diverse biomedical domains, including antiviral development against SARS-CoV-2 24 anti-virulence strategies in microbial infections, 25 oncology drug design, 26 and optimization of bioactive molecule delivery systems. 27
The simultaneous targeting of EGFR and CA-IX is a rational therapeutic strategy that addresses both proliferative signaling and hypoxia-induced metabolic adaptation. However, very few small molecules have been reported with effective dual inhibition of these targets, due to the structural and functional divergence between kinase active sites and metalloenzyme catalytic pockets.
Rationale
The 1,3,4-thiadiazole nucleus had been widely employed as a versatile scaffold in the design of anticancer agents, particularly those directed against tyrosine kinases. For instance, compound I, a sorafenib analog, acted as a potent VEGFR-2 inhibitor within this class. 28 Similarly, a 1,3,4-thiadiazole–sulfonamide hybrid (compound II) was reported as a strong anticancer candidate, demonstrating activity against both human colorectal and cervical carcinoma cells. 29 Moreover, the structural flexibility of this moiety enabled the development of compound III, which functioned as a tubulin polymerization inhibitor with remarkable anticancer potency. 30 Another thiadiazole-containing derivative, compound IV, displayed significant inhibitory effects against breast cancer cells 31 (Figure 1(a)). Some of these compounds showed good docking score and acceptable ADMET profile. Sulfonamides had been studied as anticancer scaffolds due to their ability to inhibit carbonic anhydrases, modulate protein kinases, and exert antioxidant effects, all of which contributed to suppression of tumor growth and survival. 32
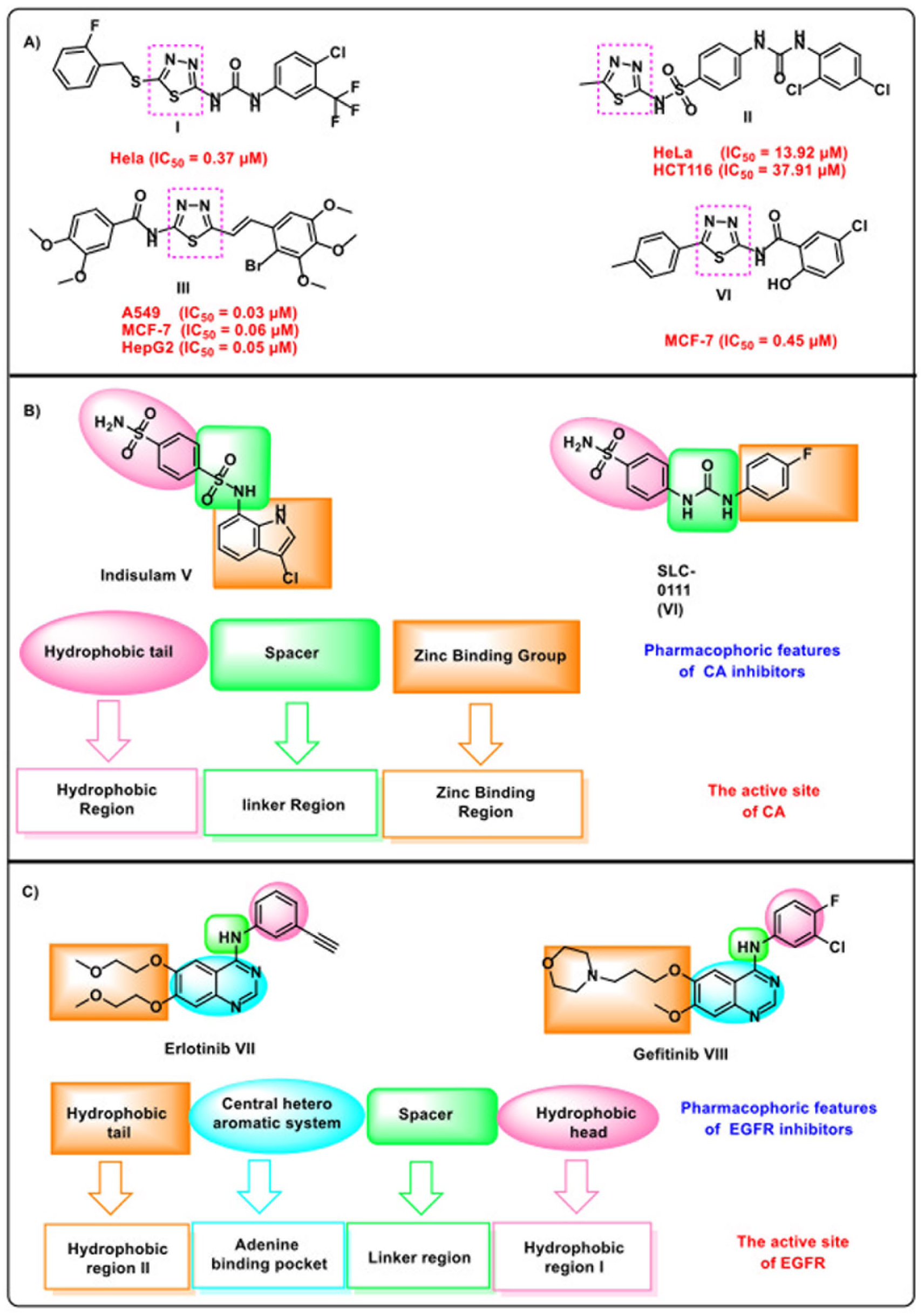
(a) Reported activities of thiadiazole derivatives against breast cancer cells. (b) pharmacophoric features of CAIs and (c) pharmacophoric features of EGFRIs.
Two carbonic anhydrase inhibitors, indisulam (V) and SLC-0111 (VI), are currently undergoing clinical trials. These compounds have demonstrated promising potential in treating solid tumors and cervical cancer.33–35 Carbonic anhydrase inhibitors (CAIs) typically share three key pharmacophoric features that enable them to effectively bind to their active site. These include a zinc-binding group, a linker moiety, and a hydrophobic tail, which interact with the zinc-binding region, spacer region, and hydrophobic pocket, respectively 36 (Figure 1(b)). Meanwhile, the FDA has approved epidermal growth factor receptor inhibitors (EGFRIs), such as erlotinib (VII) 37 and gefitinib (VIII), 38 for the treatment of various cancers, including non-small cell lung cancer and pancreatic cancer.39,40 EGFR inhibitors require four essential pharmacophoric features for optimal interaction within the active site (Figure 1(c)): (1) A planar hetero-aromatic system that fits into the adenine-binding pocket,41,42 (2) a terminal hydrophobic head that occupies hydrophobic region I. 43 (3) an NH linker group positioned in the linker region, 44 and (4) a hydrophobic tail that aligns with hydrophobic region II.45–47
In this study, we designed a novel 2,3-dihydro-1,3,4-thiadiazole and sulfonamide derivative, ASTPB, as a dual inhibitor of CA-IX and EGFR, guided by the key pharmacophoric features of both CAIs and EGFRIs. To fix the essential pharmacophoric characteristics of carbonic anhydrase, specific structural moieties were incorporated. The N-phenylbenzamide moiety functioned as the hydrophobic tail, fitting into the hydrophobic region of CA-IX. The 2-(((Z)-ethylidene)hydrazono)-2,3-dihydro-1,3,4-thiadiazole moiety served as the linker, occupying the linker region, while the benzene sulfonamide moiety acted as the zinc-binding group, targeting the zinc-binding region. Similarly, these functional groups also fixed the critical pharmacophoric features of EGFR. The 2,3-dihydro-1,3,4-thiadiazole moiety served as a heterocyclic system, fitting into the adenine-binding pocket. The benzene sulfonamide moiety functioned as the hydrophobic tail, occupying hydrophobic pocket II. The ethylidene hydrazone moiety acted as the linker, fitting into the linker region, while the N-phenylbenzamide moiety served as the hydrophobic head, occupying hydrophobic pocket I (Figure 2).

The pharmacophoric features of EGFRIs and CAIs in ASTPB.
The design of ASTPB was conceived as a proof-of-concept approach to validate the feasibility of dual CA-IX and EGFR inhibition using a thiadiazole-based scaffold before extending the study to a broader series and SAR exploration.
Results and discussions
In silico studies
Molecular docking
Molecular docking simulations serve as a robust approach for elucidating atomic-level interactions between candidate therapeutic agents and their biological targets. In this study, docking investigations centered on the compound ASTPB and its binding behavior with two critical enzymes: carbonic anhydrase-IX (PDB: 5FL4) and the epidermal growth factor receptor (EGFR, PDB: 4HJO). At first, we validated the protocol by re-docking the co-crystallized ligands into the active sites of CA-IX (PDB: 5FL4) and EGFR (PDB: 4HJO). The reproduced binding poses showed good overlap with the experimental ones, confirming the reliability of the docking workflow (Figure 3(a) and (b)). Notably, the results highlighted the essential role of the sulfonamide (SO₂NH₂) moiety as a zinc-binding group, enabling coordination with the Zn(II) ion (Figure 3(c)). Within the hCA-IX binding pocket, the SO₂NH₂ group of ASTPB established a hydrogen bond with Thr200, while the thiadiazole-2-acetyl segment formed an additional hydrogen bond with Trp9. Also, the amide bond formed other hydrogen bond with Val130. Six hydrophobic contacts were also detected, involving Leu199, Val130, Pro202, His94, and Leu91 (Figure 3(c)).

Validation of the docking protocol via superimposition of the native ligand (green) and the re-docked ligand (turquoise) within (a) CAIX active site 5FL4 (RMSD 1.15 Å) and (b) and EGFR active site 4HJO (RMSD 1.28 Å). (c) 2D view of ASTPB in the CA IX active site (PDB: 5FL4). D) 2D view of ASTPB in the EGFR active site (PDB: 4HJO). Hydrogen bonds were presented as dashed green lines, hydrophobic bonds were presented as dashed pink lines, and electrostatic bonds were presented as dashed yellow lines.
In the EGFR active site, ASTPB formed one key hydrogen bond with Met769. In addition, the molecule participated in nine hydrophobic interactions, including π-π stacking and π-alkyl contacts with Leu764, Asp831, Cys751, Leu820, Leu694, Val702, and Cys773 (Figure 3(d)).
MD simulations against EGFR
The structural stability of the EGFR–ASTPB complex was evaluated through multiple metrics derived from the 200 ns MD simulation. The root mean square deviation (RMSD) of the protein backbone exhibited an increasing trend during the initial 20 ns, reaching an average of 2.5 Å before stabilizing around 2.2 Å for the remainder of the simulation (Figure 4(a)), indicating that the system reached equilibrium and maintained a structurally stable conformation. In contrast, Figure 4(b) shows, the RMSD of the ASTPB showed higher initial fluctuations, peaking at approximately 5.5 Å within the first 10 ns, followed by a gradual decrease to around 4.5 Å, suggesting some degree of conformational flexibility while remaining bound to the protein. Analysis of global structural properties further supported the overall stability of the system. The radius of gyration (RoG) remained relatively constant at approximately 19.5 Å (Figure 4(c)), indicating minimal global structural expansion or compaction. Similarly, the solvent-accessible surface area (SASA) fluctuated around an average value of 15,250 Ų (Figure 4(d)), consistent with a stable folded state of the protein throughout the simulation. Hydrogen bonding analysis revealed that the ASTPB ligand predominantly formed a single hydrogen bond with the protein across most of the simulation time, with only transient formation of two or three hydrogen bonds observed in a minority of frames (Figure 4(e)). The root mean square fluctuation (RMSF) of Cα atoms (Figure 4(f)) indicated generally low residue-wise flexibility, except in the C-terminal region, where fluctuations reached up to 4 Å. The center-of-mass (COM) distance between the protein and the ligand decreased over the first 20 ns and subsequently stabilized at an average of around 10 Å (Figure 4(g)), reflecting a stable binding mode after initial equilibration. Taken together, these results suggest that the EGFR–ASTPB complex maintains a globally stable structure during the simulation, with notable flexibility confined primarily to the C-terminal domain, while the ASTPB remains stably associated with the protein despite displaying some conformational adaptability.

(a) RMSD values for EGFR, (b) ASTPB RMSD, (c) RoG for EGFR, (d) SASA for EGFR, (e) hydrogen bonding between ASTPB and EGFR, (f) RMSF for EGFR, (g) distance from the center of mass of ASTPB and EGFR.
In addition, MM-GBSA calculations indicated strong binding contributions from van der Waals (–57.14 kcal/mol) and electrostatic (–32.44 kcal/mol) energies (Fig. S3.2.2.1; S3.2.5), consistent with docking and inhibition profiles. ProLIF analysis revealed persistent hydrophobic contacts with Leu694, Val702, Ala719, Lys721, Met742, and Thr766 (>85% of simulation time), underscoring their role in binding stability (Fig. S3.2.3.1). PCA trajectories showed restricted motions, clustering around a stable conformational state (Fig. S3.2.3.1). FEL projections identified multiple low-energy basins with small free energy differences (0.13–0.47 kJ/mol), indicating accessible conformational states and stable dynamics (Fig. S3.2.4.1).
MD simulations against CA-IX
Over the initial 160 ns of the simulation, the CA-IX backbone exhibited stable structural fluctuations, with an average RMSD of approximately 1.5 Å, followed by a slight increase to around 2 Å for the subsequent 40 ns. Comparative structural analysis between the initial (0 ns, magenta) and late (180 ns, cyan) conformations, as highlighted in the inset of Figure 5(a), indicated that this increase was primarily attributable to increased mobility in the N-terminal region (consistent with RMSF analysis). The ligand RMSD (Figure 5(b)) suggested stable binding, characterized by relatively large RMSD values compared to the initial ligand structure. Specifically, ASTPB exhibited an increasing RMSD trend during the first 25 ns before stabilizing around 5.5 Å. Comparative structural analysis of the ligand at 0 ns (magenta sticks) and 50 ns (yellow sticks), as shown in the inset, revealed a reorientation of the outwardly facing portion of the ligand. The protein’ Radius of Gyration (RoG) (Figure 5(c)) remained relatively stable on average, with the exception of the final 40 ns, which coincided with the increased fluctuations observed in the N-terminal region. Similarly, analysis of Solvent Accessible Surface Area (SASA) values (Figure 5(d)) displayed a trend consistent with the RoG data. Furthermore, the ASTPB ligand predominantly maintained a single hydrogen bond with the protein throughout the majority of the simulation, with only a small fraction of frames exhibiting two hydrogen bonds (Figure 5(e)). The RMSF profile of the Cα atoms (Figure 5(f)) generally showed stable fluctuations, with the exception of the N-terminal region, where fluctuations reached up to 4 Å. The center-of-mass (COM) distance between the protein and the ligand (Figure 5(g)) exhibited a stable trend throughout the simulation, with an average of approximately 12 Å. Taken together, these observations suggest that the protein maintained a generally stable overall structure during the simulation, with notable flexibility in the N-terminal region, while the ASTPB ligand displayed consistent binding to the protein.

(a) RMSD for the CA-IX: CA-IX structure at 0 ns (magenta) and 180 ns (cyan); (b) ASTPB RMSD values: ASTPB structure at 0 ns (magenta) and 50 ns (yellow), (c) RoG for the CA-IX, (d) SASA for CA-IX, (e) hydrogen bonds, (f) RMSF for CA-IX, (g) distance from the center of mass of ASTPB compound and CA-IX.
Furthermore, MM-GBSA analysis indicated that van der Waals (–40.26 kcal/mol) and electrostatic (–20.06 kcal/mol) forces were the main stabilizers of the CA-IX–ASTPB complex (Fig. S3.2.2.2; S3.2.6), supporting docking and inhibition results. ProLIF revealed frequent hydrophobic interactions with Thr73, Leu91, Val130, and Leu199 (>85%) and a persistent H-bond with Thr200 (89.7%). Additional residues, including Pro75, Gln92, Val121, Leu134, and Thr201, showed weaker contacts (<85%), providing quantitative insight into binding (Fig. S3.2.3.3). PCA showed limited conformational fluctuations, indicating that ASTPB maintained a stable orientation within the active site, consistent with RMSD and H-bond analyses (Fig. S3.2.4.3; S3.2.6). FEL projections (Fig. S3.2.4.2) revealed multiple low-energy basins. In PC1–PC2, the system remained in a broad stable basin (ΔG = 0.29 kJ/mol). PC1–PC3 showed a wide basin with minimal energetic separation (ΔG = 0.09 kJ/mol). PC2–PC3 identified three basins, with transitions between them and a ΔG of 0.7 kJ/mol between the global and second minima.
The proposed dual inhibition mechanism is inferred from molecular docking, MD simulations, and cytotoxicity results, and was not experimentally validated at the protein expression or phosphorylation level. Further mechanistic studies are warranted.
DFT calculations
DFT calculations (B3LYP/6-31G+(d, p), Gaussian 09) were performed to characterize the electronic and structural properties of ASTPB (Fig. S3.3.1A; Table 1). The optimized structure showed a relatively low dipole moment, suggesting balanced charge distribution across the molecule. Frontier Molecular Orbital (FMO) analysis indicated favorable inhibitory potential, with HOMO and LUMO energies reflecting electron-donating and -accepting capacities, respectively. The small HOMO–LUMO gap (Egap) suggested efficient electron transfer and strong potential for protein interactions (Fig. S3.3.1B). TDOS analysis further supported electron-donating behavior (Fig. S3.3.1C).
The DFT calculated global reactivity parameters for ASTPB.

Charge distribution analyses highlighted potential binding sites: Mulliken charges and electrostatic potential maps revealed electron-rich oxygen atoms as likely hydrogen bond donors, while electrophilic (S14) and nucleophilic (C8) sites may mediate protein interactions (Figs. S3.3.1D–E). Negative electrostatic zones were linked to H-bond formation with polar residues, while positive and neutral regions may support interactions with negatively charged and hydrophobic residues, respectively. Based on HOMO–LUMO energies, ASTPB is a soft molecule, favoring interactions with protein targets relevant to anticancer activity.
In silico ADMET and toxicity analysis
In silico predictions demonstrated that ASTPB possesses a favorable pharmacokinetic and safety profile compared with erlotinib and acetazolamide. Very low blood–brain barrier penetration was observed, reducing risks of CNS-related side effects and making ASTPB particularly suitable for peripheral solid tumors where EGFR and CA-IX are overexpressed. Although aqueous solubility and oral absorption were predicted to be low, these limitations may be overcome through formulation approaches such as liposomes, nanoparticles, or prodrug strategies. Metabolic safety was favorable: ASTPB was predicted to be a non-inhibitor of CYP2D6, minimizing drug–drug interaction potential, while high plasma protein binding (>90%) suggested prolonged systemic circulation. Importantly, toxicity models indicated that ASTPB is non-carcinogenic, non-mutagenic, and non-toxic, in contrast to erlotinib and acetazolamide, both identified as multi-organ carcinogens, with acetazolamide also flagged for developmental toxicity.
Acute and chronic safety margins further supported ASTPB’s tolerability. The compound showed an oral LD₅₀ of 2.68 mmol/kg and a chronic LOAEL of 0.126 mmol/kg, indicating a broad therapeutic window. While acetazolamide had a higher LD₅₀, its greater chronic toxicity (LOAEL = 0.23 mmol/kg) suggested accumulation-related risks; erlotinib displayed the narrowest safety margin. Dermal and ocular assessments predicted ASTPB as non-irritant to skin and only slightly irritating to eyes, supporting its suitability for parenteral or topical administration.
Overall, ASTPB combines favorable ADME properties, minimal toxicity, and a broad safety margin. Its low CNS penetration, absence of CYP inhibition liabilities, and non-carcinogenic profile make it a promising candidate for systemic anticancer therapy, warranting further preclinical evaluation and formulation optimization. However, although the in silico ADMET predictions provided useful preliminary insights into the pharmacokinetic and safety profile of ASTPB, these results remain computational estimates. Experimental validation through in vitro and in vivo ADMET assays will be essential to confirm these findings
Synthesis and characterization of ASTPB
ASTPB was synthesized as outlined in Scheme 1. Intermediate 4 was obtained by treating pentane-2,4-dione 3 with sulfuryl dichloride in anhydrous ethoxyethane under ice–salt cooling. Reaction of intermediate 4 with 4-sulfamoylbenzenediazonium chloride 2 (generated by diazotizing sulfanilamide 1 with NaNO₂/HCl at 0–5 °C) afforded hydrazonoyl chloride 5. Separately, p-aminoacetophenone 6 was acylated with benzoyl chloride in DMF/TEA to yield N-(4-acetylphenyl)benzamide 8, which upon condensation with methyl hydrazinecarbodithioate 12 furnished intermediate 13. Final cyclocondensation of 13 with hydrazonoyl chloride 5 in EtOH/TEA produced ASTPB in high yield. ASTPB was isolated as a yellow crystalline solid (mp 205–207 °C). Its IR spectrum showed NH and SO₂NH₂ stretching at 3361 and 3257 cm⁻¹, and strong carbonyl bands at 1690 and 1655 cm⁻¹. The ¹H NMR spectrum displayed a singlet at δ 10.44 ppm (NH), aromatic multiplets at δ 7.83–8.00 ppm, and singlets at δ 2.28 and 2.43 ppm (methyl groups). The ¹³C NMR spectrum showed a carbonyl resonance at δ 190.43 ppm and signals at δ 166.27, 164.31, and 161.34 ppm, consistent with amide and thiadiazole carbons. Mass spectrometry confirmed the molecular ion peak at m/z 534, matching the calculated molecular weight. Elemental analysis (C, 56.35; H, 4.23; N, 15.86%) was in excellent agreement with theoretical values (C, 56.17; H, 4.15; N, 15.72%). These data confirmed the successful synthesis and purity of ASTPB.

Synthesis pathway of ASTPB.
In vitro examinations
Enzyme assay
ASTPB demonstrated potent dual inhibition of CA-IX and EGFR, with IC₅₀ values of 0.046 ± 0.007 µM and 0.059 ± 0.009 µM, respectively. Its CA-IX inhibition closely matched acetazolamide (0.039 ± 0.005 µM), while its EGFR inhibition was slightly weaker than erlotinib (0.027 µM). Importantly, unlike the single-target reference drugs, ASTPB simultaneously inhibited both CA-IX and EGFR—two pathways frequently co-expressed in hypoxic, aggressive tumors. This dual-targeting property may yield synergistic therapeutic effects, enhancing efficacy and potentially overcoming resistance mechanisms.
Cytotoxicity
The cytotoxicity assessment of ASTPB across a panel of human cancer cell lines (HePG-2 (hepatocellular carcinoma), MCF-7 (breast cancer), HCT-116 (colon carcinoma), PC-3 (prostate cancer), and MDA-MB-231 (triple-negative breast cancer)) revealed a moderate to good anticancer profile, particularly in MDA-MB-231 (IC₅₀ = 18.15 ± 1.4 µM) and MCF-7 (25.39 ± 1.7 µM) cells (Table 2). These findings are notable considering the structural design of ASTPB as a dual inhibitor of CA-IX and EGFR, both of which are overexpressed in aggressive and metastatic tumor types, including TNBC. Compared to the reference drug doxorubicin (DOX), ASTPB exhibits higher IC₅₀ values, indicating lower intrinsic cytotoxicity. However, this may reflect a targeted mechanism of action rather than broad-spectrum cytotoxicity, which is often associated with adverse effects in healthy tissues. This is further supported by the compound’s relatively high IC₅₀ in WI-38 normal fibroblasts (72.46 ± 3.6 µM), suggesting a favorable therapeutic window and reduced off-target toxicity. The safety profile of ASTPB aligns with the rational design strategy of dual inhibition to selectively modulate tumor-associated targets, rather than indiscriminately damaging both cancerous and normal cells.
In vitro cytotoxicity (IC₅₀, µM) of ASTPB in normal and cancer cell lines.

Data are presented as the mean ± standard error of the mean (SEM) from three separate experiments
Interestingly, among the tested lines, MDA-MB-231 showed the highest sensitivity to ASTPB, consistent with literature reports highlighting the role of EGFR overexpression and CA-IX upregulation in hypoxic, basal-like breast cancers. The compound’s efficacy in HCT-116 and HePG-2 cells (IC₅₀ = 31.83 and 39.86 µM, respectively) also aligns with the involvement of EGFR/CA-IX in colorectal and hepatic tumors, suggesting a broad-spectrum dual-target engagement across tumor types with known resistance mechanisms. Overall, while ASTPB is not as cytotoxic as doxorubicin in vitro, its selective and mechanistically-driven activity, lower toxicity in normal cells, and broad antitumor potential present it as a lead candidate for further preclinical development.
Cell cycle analysis
Cell cycle analysis in MDA-MB-231 cells treated with ASTPB (Fig. S.2.4.1 and Table 3) revealed a significant G0/G1 phase arrest, with a marked increase in the G0/G1 population from 37.45% (control) to 63.56%. This was accompanied by a notable reduction in both S-phase (from 32.72% to 17.46%) and G2/M phase (from 29.83% to 18.98%), suggesting that ASTPB exerts a cytostatic effect by halting cell cycle progression at the G1 checkpoint. This arrest pattern is mechanistically consistent with EGFR inhibition, as EGFR is a known regulator of cell proliferation via RAS/RAF/MEK/ERK and PI3K/AKT signaling cascades, which converge on cyclin-dependent kinases (CDKs) controlling G1/S transition.48,49 By attenuating EGFR signaling, ASTPB likely downregulates cyclin D1 and CDK4/6 activity, resulting in reduced phosphorylation of the retinoblastoma protein and thereby enforcing G1 arrest. In addition, CA-IX inhibition may synergize with this mechanism under hypoxic conditions commonly present in TNBC tumors. CA-IX helps maintain pH homeostasis in hypoxic cells, which is essential for metabolic adaptation and cell cycle progression. Its inhibition may disrupt intracellular pH, leading to metabolic stress and impaired DNA synthesis during S-phase, further contributing to the observed cell cycle blockade.
Cell cycle distribution of MDA-MB-231 cells treated with ASTPB.

Significant P value significant P value < 0.05 & by using One-way ANOVA followed by Tukey’s post hoc multiple comparison tests.
Collectively, these data provide functional confirmation of ASTPB’s ability to interfere with cell proliferation through dual-targeted signaling disruption and support its development as a novel G1-phase-specific inhibitor in aggressive EGFR/CA-IX-overexpressing tumors such as MDA-MB-231.
Apoptosis and necrosis rates of MDA-MB-231
Annexin V/PI staining of MDA-MB-231 cells treated with ASTPB (Fig. S.2.5.1 and Table 4) revealed a substantial increase in apoptosis, with early apoptotic cells rising to 76.73%, compared to just 0.10% in untreated controls. Minimal levels of late apoptosis (5.67%) and necrosis (0.17%) were observed, while the proportion of viable cells dropped sharply from 98.46% in controls to 17.42%. These results indicate that ASTPB predominantly induces early-stage programmed cell death, rather than triggering necrosis or late apoptosis. This apoptotic response is consistent with the compound’s ability to simultaneously inhibit CA-IX and EGFR, two key mediators of apoptosis resistance. EGFR signaling supports cell survival by activating PI3K/AKT and MAPK/ERK pathways, which, in turn, regulate anti-apoptotic proteins like Bcl-2 and surviving. 50 By targeting EGFR, ASTPB likely suppresses these pro-survival signals, promoting mitochondrial destabilization and activation of the intrinsic apoptotic pathway.
Apoptosis profile of MDA-MB-231 cells after treatment with ASTPB.

Significant P value significant P value < 0.05 & by using One-way ANOVA followed by Tukey’s post hoc multiple comparison tests.
At the same time, inhibition of CA-IX under hypoxic or acidic tumor conditions may further promote apoptosis by disrupting pH balance, impairing ion regulation, and enhancing oxidative and metabolic stress. 51 These effects may facilitate mitochondrial membrane permeabilization and cytochrome c release, boosting caspase cascade activation in response to dual inhibition.
The predominance of early apoptosis, coupled with minimal necrosis, suggests a controlled, non-inflammatory mode of cell death—an advantageous trait that could minimize damage to surrounding healthy tissues and reduce unwanted immune responses.
When considered alongside the observed G0/G1 phase cell cycle arrest, these findings support a mechanism in which ASTPB initially halts tumor cell proliferation and subsequently triggers a strong apoptotic response. This dual action underscores the compound’s rational design and potential as a targeted anticancer agent capable of overcoming resistance in aggressive tumor types
Apoptotic protein markers
The pro-apoptotic activity of ASTPB was further validated at the molecular level by assessing the expression of key proteins involved in the intrinsic (mitochondrial) apoptotic pathway (Table 5). Upon treatment of MDA-MB-231 cells with ASTPB, a significant increase in pro-apoptotic markers was observed (Bax levels rose to 211.85 ng/mL (3.7-fold increase), and caspase-3 to 210.36 ng/mL (also 3.7-fold)) while the anti-apoptotic protein Bcl-2 decreased sharply to 2.64 ng/mL (1.97 -fold) relative to untreated controls. This substantial shift in the Bax/Bcl-2 ratio is a pivotal event leading to mitochondrial outer membrane permeabilization. Bax oligomerization facilitates cytochrome c release into the cytosol, which in turn activates the caspase cascade. The robust upregulation of caspase-3, a key executioner in apoptosis, strongly supports the conclusion that ASTPB initiates cell death through the mitochondrial pathway.
Levels of apoptotic markers in MDA-MB-231 cells treated with ASTPB.

Significant P value significant P value < 0.05 & by using One-way ANOVA followed by Tukey’s post hoc multiple comparison tests.
Mechanistically, this apoptotic cascade aligns with ASTPB’s dual-targeting approach. First, inhibition of EGFR suppresses the PI3K/AKT signaling axis, which ordinarily upregulates Bcl-2 and inhibits Bax activation, thereby removing key anti-apoptotic signals. In addition, the inhibition of CA-IX disrupts pH regulation under hypoxic conditions, intensifying metabolic and oxidative stress that exacerbates mitochondrial damage and promotes caspase activation. These molecular effects reinforce earlier findings (including a pronounced early apoptotic response (76.7%), G0/G1 cell cycle arrest, and moderate cytotoxicity in TNBC cells) indicating that ASTPB activates a well-coordinated apoptotic program through both upstream signaling interference and downstream mitochondrial effector activation.
Statistical analysis section
All data were analyzed using GraphPad Prism 6 (GraphPad, San Diego, CA, USA) and are presented as mean ± SD from three independent experiments (n = 3). Statistical significance was assessed by one-way ANOVA with Tukey’s post hoc test, with P < 0.05 considered significant.
Novelty
This work introduces ASTPB as the first rationally designed thiadiazole–sulfonamide hybrid that incorporates pharmacophoric features of both CA-IX and EGFR inhibitors into a single scaffold, overcoming the structural divergence that has limited previous dual-target efforts. Synthesized and fully characterized as a novel molecular entity, ASTPB demonstrated potent dual inhibition with submicromolar IC₅₀ values against both enzymes, in contrast to the single-target activity of erlotinib and acetazolamide. Importantly, ASTPB showed selective cytotoxicity toward triple-negative breast cancer cells (MDA-MB-231) while sparing normal fibroblasts (WI-38), highlighting its therapeutic selectivity compared to broadly toxic agents like doxorubicin. Mechanistic studies revealed additional novelty, with ASTPB inducing G0/G1 arrest, triggering apoptosis (76.7%), and modulating apoptotic protein markers (Bax and caspase-3 upregulated; Bcl-2 downregulated), findings not previously reported for dual CA-IX/EGFR inhibitors. Finally, the originality and rigor of this study were reinforced by the integration of pharmacophore modeling, docking, long-timescale MD simulations, DFT analysis, ADMET predictions, chemical synthesis, enzymatic inhibition, cytotoxicity assays, cell cycle analysis, apoptosis, and protein marker evaluations, together providing comprehensive multi-level validation of ASTPB as a promising anticancer candidate.
Limitation
While this study primarily employed molecular docking, MD simulations, and DFT analyses to guide the design and predict the binding behavior of ASTPB, we also performed in vitro assays (enzyme inhibition, cytotoxicity) that validated its biological activity and supported the computational predictions. However, no in vivo pharmacokinetic, efficacy, or toxicity studies were conducted to confirm the biological relevance of ASTPB. Future work will focus on evaluating the compound’s in vivo performance and safety profile to further validate its therapeutic potential.
Conclusion
In this study, we designed, synthesized, and evaluated ASTPB, a novel thiadiazole–sulfonamide derivative developed as a dual CA-IX/EGFR inhibitor. ASTPB exhibited potent enzyme inhibition (IC₅₀ = 0.046 ± 0.007 µM for CA-IX; 0.059 ± 0.009 µM for EGFR), comparable to standard reference drugs. In cytotoxicity assays, it selectively suppressed triple-negative breast cancer cell proliferation (MDA-MB-231, IC₅₀ = 18.15 µM) while showing reduced toxicity toward normal fibroblasts (WI-38, IC₅₀ = 72.46 µM), reflecting a favorable therapeutic index. Mechanistic studies revealed G0/G1 arrest (63.56% vs 37.45% in control) and induction of apoptosis in 76.7% of treated cells, accompanied by Bax and caspase-3 upregulation (3.7-fold increase) and Bcl-2 downregulation (1.97-fold decrease), consistent with activation of the intrinsic apoptotic pathway. Together, these findings establish ASTPB as a promising lead for multi-targeted anticancer therapy, particularly against aggressive, hypoxia-adapted tumors co-expressing CA-IX and EGFR. Future work will focus on synthesizing ASTPB analogs to develop structure–activity relationships (SAR) for potency and selectivity optimization, alongside pharmacokinetic studies and in vivo validation to assess efficacy, safety, and bioavailability. Broader evaluation across CA-IX/EGFR co-expressing cancer models, as well as rescue experiments using target overexpression or knockdown, will further confirm that the observed effects are specifically mediated through dual inhibition.
Experimental
Chemistry
General method for the synthesis of ASTPB: N-(4-((E)-1-(((Z)-5-Acetyl-3-(4-sulfamoylphenyl)-1,3,4-thiadiazol-2(3H)-ylidene)hydrazono)ethyl)phenyl)benzamide
The target compound, ASTPB, was obtained through a cyclocondensation reaction between intermediate 13 and (Z)-2-oxo-N-(4-sulfamoylphenyl)propanehydrazonoyl chloride 5 in a 1:1 molar ratio. The reaction was carried out in 25 mL of ethanol with triethylamine (TEA) as a catalytic base. The mixture was magnetically stirred and refluxed for 6 h to ensure complete conversion. After cooling to room temperature, the resulting precipitate was collected by filtration, washed with absolute ethanol, air-dried, and recrystallized from a suitable solvent to yield the final product ASTPB in high purity and yield.
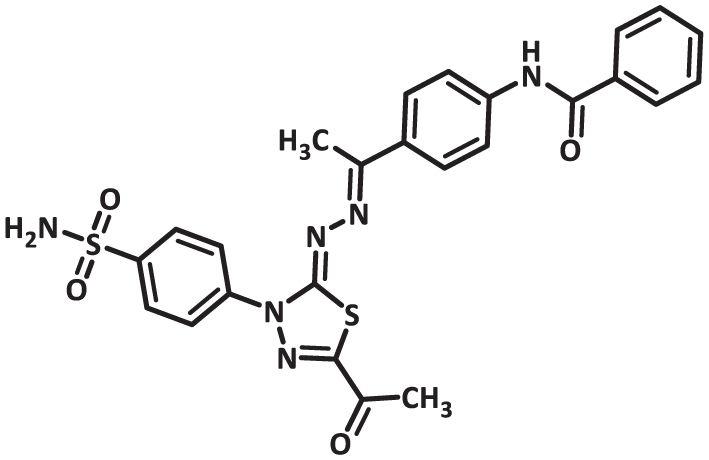
Yellow powder (Yield 78%), melting point 205–207 °C; IR (KBr, υmax cm-1): FTIR (v max, cm-1): 3361, 3257, 3106, 3011, 1690, 1655, 1601, 1528; 1H NMR (500 MHz, DMSO-d6) δ 10.44 (s, 1H, NH), 8.00–7.83 (m, 9H, ArH), 7.57 (t, J = 7.3 Hz, 2H, ArH), 7.54–7.48 (m, 2H, ArH), 7.46 (s, 2H, SO2NH2), 2.43 (s, 3H, CH3), 2.28 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-D6) δ 190.43, 166.27, 164.31, 161.34, 158.06, 152.05, 142.37, 141.19, 135.28, 133.68, 132.69, 132.29, 128.98, 128.28, 127.57, 127.37, 122.23, 25.64, 15.07; MS (m/z) = 534 [%]:[M+, (45.55%)], Anal. Calcd for C25H22N6O4S2 (534.61): C, 56.17; H, 4.15; N, 15.72; Found: C, 56.35; H, 4.23; N, 15.86%.
Computational studies
Molecular docking
The crystal structures of CA-IX, and EGFR were retrieved from the Protein Data Bank. The docking workflow involved ligand optimization, binding site specification, and pose scoring based on binding energy values. 52 Further details, including validation steps, are available in the Supplementary Data. For CA-IX, we employed PDB ID: 5FL4, which represented a high-resolution crystal structure of human carbonic anhydrase-IX in complex with a sulfonamide, thus providing a reliable template for studying the zinc-binding pocket. For EGFR, PDB ID: 4HJO in complex with a erlotinib was employed to investigate interactions at the binding site.
Molecular dynamics (MD) simulations
To evaluate the structural stability of the ASTPB-protein complexes, 200 ns MD simulations were carried out using GROMACS 2021.53,54 System preparation involved CHARMM-GUI and solvation in a TIP3P water box under periodic boundary conditions, with the CHARMM36 force field applied. Analysis included RMSD, RMSF, hydrogen bonding, and radius of gyration metrics. Binding free energies were calculated using MM-GBSA via the gmx_MMPBSA tool.55,56 In addition, PCA was used to assess conformational changes. 57 Full computational parameters are included in the Supplementary Data.
Density functional theory (DFT) calculations
The electronic structure and chemical reactivity of ASTPB were investigated through DFT studies using Gaussian 09. Geometry optimization was performed at the B3LYP/6-31+G(d, p) level. 58 Key electronic parameters such as HOMO-LUMO gap, dipole moment, and molecular electrostatic potential (MEP) maps were calculated to evaluate the molecule’s stability and reactivity toward biological systems. Additional information is provided in the Supplementary Data.
ADMET and toxicity prediction
The pharmacokinetics and toxicological profile of the synthesized thiadiazole derivative were computationally predicted,59,60 detailed in the Supplementary Data.
In vitro evaluation
Cytotoxicity assay
The anticancer activity of ASTPB was assessed using the MTT assay against multiple human cancer cell lines: 61 HePG-2 (liver), MCF-7 and MDA-MB-231 (breast), HCT-116 (colon), and PC-3 (prostate). Normal human fibroblast cells (WI-38) were included to determine selectivity and cytotoxic index. Cells were exposed to varying concentrations of ASTPB for 48 h, and viability was measured by mitochondrial activity. IC₅₀ values were derived from triplicate experiments. Protocol details are available in the Supplementary Data.
Enzymatic inhibition assays
The inhibitory potential of ASTPB against CA-IX and EGFR was confirmed via in vitro enzyme inhibition assays. 62 Serial dilutions (0.3–1000 nM) were used to construct dose-response curves and determine IC₅₀ values. Acetazolamide and erlotinib were employed as reference inhibitors for CA and EGFR, respectively. Experimental conditions and analysis methods are described in the Supplementary Data.
Apoptosis and cell cycle analysis by flow cytometry
Flow cytometry was used to analyze the effects of ASTPB on cell cycle phases and apoptosis induction in MDA-MB-231 cells. 63 Cell cycle progression was assessed using propidium iodide staining, while apoptosis was determined using Annexin V-FITC/PI dual staining. Quantitative analysis differentiated between viable, early and late apoptotic, and necrotic cells. Detailed experimental protocols are presented in the Supplementary Data.
Apoptosis-related protein expression
Enzyme-linked immunosorbent assay (ELISA) assays were used to quantify expression levels of Bax, Bcl-2, and caspase-3 proteins in MDA-MB-231 cells following treatment. Data were normalized and expressed as fold change relative to untreated controls. Full methodologies are described in the Supplementary Data.
Supplemental Material
sj-pdf-1-chl-10.1177_17475198251400403 – Supplemental material for A novel thiadiazole-based dual inhibitor of carbonic anhydrase-IX and epidermal growth factor receptor targeting cancer: A combined in silico and in vitro approach
Supplemental material, sj-pdf-1-chl-10.1177_17475198251400403 for A novel thiadiazole-based dual inhibitor of carbonic anhydrase-IX and epidermal growth factor receptor targeting cancer: A combined in silico and in vitro approach by Eslam B Elkaeed, Hazem Elkady, Walid E Elgammal, Hazem A Mahdy, Dalal Z Husein, Ibrahim M Ibrahim, Hanan A Al-ghulikah, Ibrahim H Eissa and Ahmed M Metwaly in Journal of Chemical Research
Footnotes
Acknowledgements
The authors thank Research Center at AlMaarefa University for supporting this work.
Authorship contributions
E.B.E.: Supervision, Resources, Formal analysis, editing; H.E.: software, writing—review & editing; W.E.E.: methodology; H.A.M.: writing—original draft; D.Z.H.: software, methodology; I.M.I.: software; H.A.A.-g.: funding acquisition, writing—review & editing; I.H.E.: project administration, supervision; A.M.M.: Writing—review & editing, supervision.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R95), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The data that support the findings of this study are enclosed in the manuscript and the supplementary materials.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.