
Abstract
4-nitro-3,5-dimethylpyridine-N-oxide is a key intermediate in the preparation of esomeprazole, which involves oxidation followed by nitration reactions. However, the conventional batch reactor method necessitates use of excess peroxide, and nitric acid/sulfuric acid mixture which faces serious safety and environmental issues. Herein, we report efficient production of 4-nitro-3,5-dimethylpyridine-N-oxide using the microchannel technology which not only increases the yield but also significantly reduces safety risks and waste output.
Introduction
Contemporary epidemiological investigations demonstrate a marked escalation in the incidence of acid-related peptic ulcers and associated gastrointestinal disorders, principally attributed to occupational stressors and dietary pattern alterations.1,2 Clinical management of these pathologies centers on gastric acid suppression, with proton pump inhibitors (PPIs) such as esomeprazole.3–6
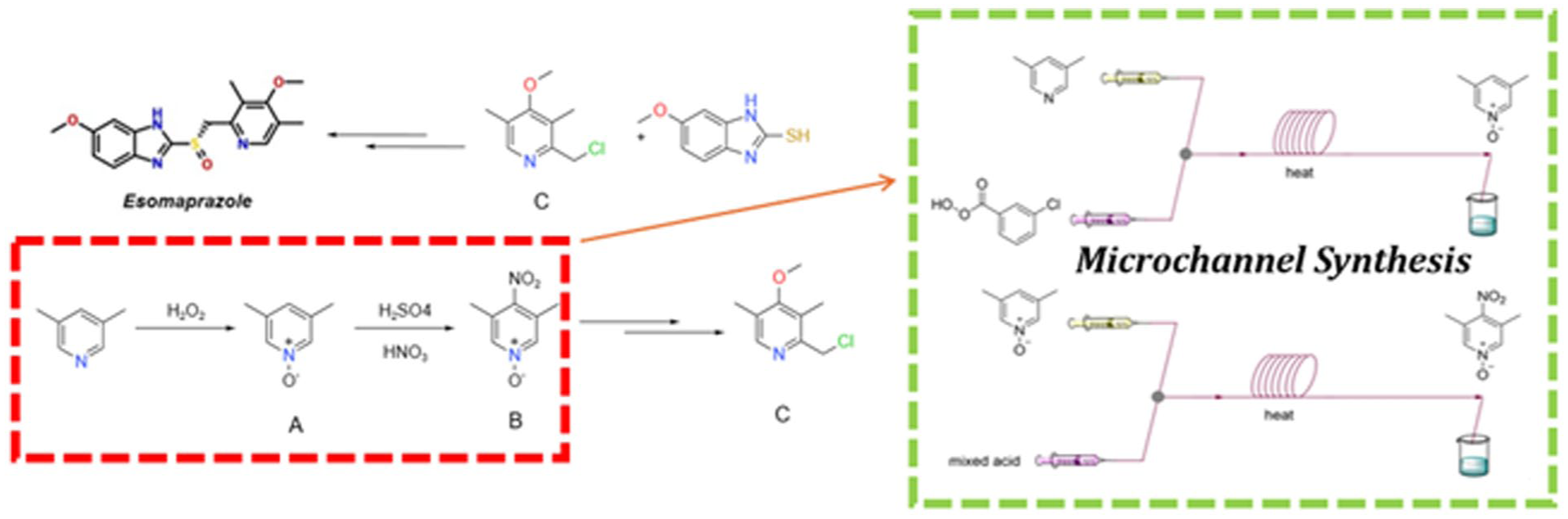
4-nitro-3,5-dimethylpyridine-N-oxide (B) serves as a pivotal synthetic intermediate in the preparation of 2-(chloromethyl)-4-methoxy-3,5-dimethylpyridine (C), a critical pharmaceutical building block for esomeprazole (Scheme 1). Its synthesis involves highly exothermic oxidation and nitration reactions that demand stringent safety protocols.7–9 This conventional batch reactor production requires the use of high-concentrated hydrogen peroxide and concentrated nitric acid/sulfuric acid at high temperature (Scheme 1), which face a significant risk of explosion and equipment corrosion problems.10–12 Meanwhile, the large amount of nitrogen-containing wastewater produced by this method increases the waste-treatment cost.13,14 Therefore, in terms of production safety and wastewater treatment, this technical route should be further improved.

The oxidation and nitration steps of 3,5-dimethylpyridine.
Microchannel reactors exhibit significantly reduced reaction zone dimensions and enhanced surface-to-volume ratios, enabling rapid heat transfer that substantially improves reaction efficiency while minimizing energy and raw material consumption (Figure 1). The microscale reaction volumes inherently mitigate risks associated with hazardous chemical processes by eliminating requirements for extreme temperature/pressure conditions.15,16 These characteristics render the technology particularly suitable for handling dangerous and resource-consuming industrial reactions. Compared with conventional batch reactors, microchannel systems achieve superior process control through advanced terminal monitoring, effectively suppressing side reactions while enabling precise automation of reaction parameters. Owing to these pronounced advantages, microchannel reactors have been widely implemented in organic transformations such as Suzuki-Miyaura cross-couplings, Diels-Alder cycloadditions, and desulfurization processes of organic compounds,17–20 and pharmaceutical synthesis.21–24
Considering the high safety profiles and conversion efficiency of microchannel technique for these hazardous transformations, we herein report on efficient production of 4-nitro-3,5-dimethylpyridine-N-oxide using microchannel reactors (Scheme 2). Through precise regulation of reaction parameters including temperature, residence time and feeding stoichiometry, the associated risks with the traditional batch reactor are substantially mitigated. Under the optimized condition, the yield of 4-nitro-3,5-dimethylpyridine-N-oxide using microchannel reactor was increased from 62.8% over batch reactor to 67.3%, while significantly reducing safety risks and waste output.

Schematic diagram of microchannel reaction in this work.
Experimental
Materials
Unless stated otherwise, the reagents were used as received without further purification. 3,5-dimethylpyridine (⩾98%), acetic acid, hydrogen peroxide aqueous solution (H2O2, 30%), concentrated nitric acid (HNO3, 65–68%), fuming nitric acid (HNO3, 90–97.5%), concentrated sulfuric acid (H2SO4, 98%), m-chloroperbenzoic acid (m-CPBA, 85%), dichloromethane (DCM, 99.5%).
Conventional batch reactor synthesis
A 500 mL three-necked round-bottom flask was charged with 65.7 g of 3,5-dimethylpyridine and heated to 50°C under mechanical stirring. A 30% aqueous H2O2 solution (55.5 g, containing 16.65 g of H2O2) and acetic acid was introduced dropwise over 3 h, during which the reaction temperature was gradually elevated to 80°C. The mixture was maintained at 80°C for 3 h with continuous agitation. Upon completion of the initial reaction phase, the system was cooled to 50°C, followed by controlled addition of a second 30% aqueous H2O2 solution (33.3 g, containing 9.99 g of H2O2) over 1 h. The reaction vessel was subsequently heated to 100 °C and held isothermally for 6 h. Reaction progression was monitored at 30-min intervals by reversed-phase HPLC (C18 column, 254 nm detection, acetonitrile/water mobile phase 70:30 v/v).
After completion of the reaction, the solvent was removed by reduced-pressure distillation, and the resulting solid (3,5-dimethylpyridine-N-oxide referred as A) was directly subjected to the nitration step. Under vigorous stirring, 220 mL of mixed acid (prepared from concentrated nitric acid and concentrated sulfuric acid in a 2:1 v/v ratio) was added dropwise over 30 min. The reaction system was then heated to 90°C and maintained for 5 h with reaction progress monitored by HPLC. Upon completion, the mixture was cooled to room temperature and slowly poured into ice-water (500 mL) with mechanical stirring, while maintaining the system temperature at 0°C. The pH was adjusted to weakly acidic conditions (pH 5-6) through controlled ammonia gas introduction. The precipitated product (4-nitro-3,5-dimethylpyridine-N-oxide, referred as B) was collected by filtration, dried under reduced pressure to constant weight.
Microchannel reactors synthesis
3,5-dimethylpyridine (1.0 g, 0.0093 mol) was dissolved in 20 mL of DCM and delivered via a syringe pump at a constant flow rate (X1 mL min–1) into the reaction system. Concurrently, 85% m-CPBA (2.36 g, 0.012 mol) dissolved in 30 mL of DCM was introduced through a second syringe pump (flow rate Y1 mL min–1). The mixed reactants were heated to T1 °C and maintained for a specific residence time of t1 min in a polytetrafluoroethylene (PTFE) reaction tube (3 mL volume, 1.0 mm inner diameter). The resultant mixture was collected and then purified by recrystallization.
Compound A (1.0 g, 0.0081 mol) was dissolved in 50 mL of concentrated sulfuric acid and delivered via a syringe pump at a constant flow rate (X2 mL min–1) into the microreactor system. Simultaneously, an equivalent volume of pre-mixed nitrating acid solution (prepared from 50 mL fuming nitric acid and 50 mL concentrated sulfuric acid) was introduced through a second syringe pump (flow rate Y2 mL min–1). The combined streams underwent nitration in a PTFE reaction tube (10 mL volume, 1.0 mm inner diameter) heated to T2 °C with controlled residence time (t2 min). Upon reaction completion, the mixture was quenched with ice-water (200 mL) and extracted with DCM. The organic phase was collected in 10 mL centrifuge tubes, with 1 μL samples withdrawn from the organic phase for HPLC analysis.
Compounds A and B were characterized by 1H NMR, ESI-MS and UV-Vis spectra (Supplemental Figures S1 and S2).
Results and discussion
Conventional batch synthesis
In the conventional batch reactor synthesis, the oxidation step necessitated approximately 14 h to achieve complete conversion of 3,5-dimethylpyridine, entailing meticulous thermal regulation and gradual reagent addition. Notably, the use of concentrated hydrogen peroxide introduced substantial exothermicity, exacerbating thermal management challenges and compromising process safety, which critically restricts its industrial scalability.
The followed 7-h nitration process yielded 47.2 g of compound B (48.2% of isolated yield) with 99.4% HPLC purity. However, this step employed concentrated mixed acid (H2SO4/HNO3), followed by pH modulation via gaseous ammonia neutralization, generating non-recyclable nitrogen-contaminated effluent and significant resource inefficiency. Collectively, the conventional batch production exhibits substantial technical and environmental barriers to industrial implementation.
Microchannel reactor synthesis
The oxidation step in the microchannel reactor synthesis is influenced by the reaction temperature, residence time, and feed stoichiometry. At first, temperature and residence time for microchannel reactor synthesis were optimized. Systematic temperature modulation from 15 to 40°C revealed that the isolated yield of compound A first increased, and then dropped with the increasing temperature (Figure 1(a)). Maximum conversion efficiency (100.0% yield, HPLC purity >99.9%) was achieved at 35°C. This nonlinear thermal response profile suggests competing kinetic regimes: Below 35°C the reactants are not fully activated, while above 35°C thermal decomposition pathways become progressively significant.25,26

(a) Yield of compound A at different temperatures and (b) yield of compound A at different residence time.
The retention time was modulated through proportional adjustments to the feed flow rate while maintaining constant ratios between reactants. The yield of compound A increased with the residence time increasing from 10 to 20 min, and slightly decreased with further increasing residence time (Figure 1(b)). Maximum yield of compound A (100%) was achieved at 20 min of residence time under optimized conditions (3 mL of reaction volume, 1.5 eq m-CPBA, t1 = 20 min, X1 = 0.032 mL min–1, Y1 = 0.118 mL min–1, T1 = 35°C). However, extended residence times beyond this optimum resulted in progressive yield deterioration from 100% to 97.8% at 25 min and 97.5% at 30 min.
Feed stoichiometry was precisely controlled through coordinated adjustment of reactant flow rates. The effect of the ratio and the flow rate on the yield is summarized in Table 1. Systematic evaluation revealed a linear correlation between the oxidant-to-substrate ratio (1.2-1.5 equiv.) and product yield, with compound A yield increasing from 62.4% to 100.0%. Complete conversion efficiency was achieved at 1.5 equivalents, corresponding to 0.032 mL min–1 of X1 and 0.118 mL min–1of Y1. However, further stoichiometric excess (1.6 equiv.) caused a 1.8% yield reduction, attributable to competing side reactions induced by reagent overloading.27,28
Effect of reactant ratio and feed flow rate on the yield of A.

The conventional batch oxidation of 3,5-dimethylpyridine necessitates hazardous handling of concentrated hydrogen peroxide under prolonged thermal activation (14 h in total). In contrast, the continuous-flow microreactor system achieved quantitative conversion (100% yield by HPLC) using 1.5 equivalents of m-CPBA under precisely controlled conditions (35°C, 20 min of residence time). This represents a significant reduction in thermal energy input compared with batch reactors, while eliminating the explosive risks associated with concentrated H2O2 storage and handling.
The nitration step in the microchannel reactor synthesis was also optimized in terms of reaction temperature, feed stoichiometry, residence time, and channel diameter. The nitration reaction yield exhibited a characteristic volcano-shaped temperature dependence (Figure 2), peaking at 120°C (isolated yield of 62.93%). Suboptimal temperatures below 100°C resulted in incomplete conversion (53.87%), while thermal decomposition dominated at 130°C (yield decline to 56.91%).29–31

Effect of temperature on yield of B.
The acid-to-substrate ratio was modulated by adjusting the feed flow rates of mixed acid and compound A solution while maintaining other conditions (10 mL of reaction volume, T2 = 120°C, t2 = 20 min). Increasing the molar ratio from 5:1 to 11:1 led to a yield reduction from 59.37% to 39.56% (Table 2).
Effect of reactant ratio and feed flow rate on the yield of B.

The residence time dependence of compound B formation was systematically investigated under otherwise constant reaction conditions (10 mL reactor volume, T2 = 120°C, 5:1 acid-to-substrate ratio). As illustrated in Figure 3(a), the isolated yield increased from 50.07% at 5 min to 67.32% at 15 min, followed by a decline to 59.37% at 20 min. Initial yield enhancement corresponds to complete nitration, while prolonged exposure induces side reactions.32–34

(a) Yield of compound B at different residence time and (b) yield of compound B at different inner diameter.
The channel diameter of microreactors exhibits significant impacts on reaction yield under constant optimal reaction conditions established in previous studies (10 mL reactor volume, T2 = 120°C, 5:1 acid-to-substrate ratio, t2 = 15 min).35–37 When increasing the diameter from 0.5 to 1.0 mm, the yield remarkably improved from 42.22% to 67.32%. However, further expansion to 1.5 mm resulted in a yield reduction to 54.69%, and continued diameter enlargement to 3.0 mm caused a more pronounced decline to 35.56% (Figure 3(b)). This nonlinear correlation demonstrates the critical role of dimensional optimization in microreactor design. The observed yield profile suggests the existence of an optimal channel geometry that balances mass transfer enhancement against thermal management requirements.
With the optimal parameters for microchannel reactors, microchannel synthesis of compound B was further repeated 5 times to support the reliability of this technique (Supplemental Table S1). By comparison between batch reactor and microchannel reactor, it can be seen that microchannel reactor synthesis significantly improves the reaction efficiency in terms of energy input, yield of product and safety control. Moreover, a microchannel reactor is also very suitable for large-scale continuous chemical processes not only because of its superior mass and heat transfer, ultra-high throughput, and the ability to withstand high temperatures and pressures, but also its precise control, and straightforward mechanical assembly that simplifies maintenance. Finally, it is worth noting that the microchannel reactor synthesis of compounds A and B from 3,5-dimethylpyridine is a very cost-efficient route in this case because the starting material of 3,5-dimethylpyridine can be cheaply obtained in our company that is actually a side-product in the other chemical process. In order to clearly demonstrate the advantages of microchannel technology, a detailed quantitative comparison between the batch and microchannel reactors in terms of waste production and energy consumption has been made in Supplemental Table S2.
Although the laboratory-scale results demonstrate clear advantages of the microchannel reactor for the synthesis of 4-nitro-3,5-dimethylpyridine-N-oxide, the pathway to industrial implementation requires addressing several key engineering challenges. The most frequently cited concerns for continuous-flow microreactors are channel clogging and maintenance. On the one hand, solid formation or precipitate deposition, potentially from by-products or insufficient solubility, can lead to channel blockage during microchannel synthesis. This is a common concern for nitration reactions where insoluble intermediates or products may form. However, several strategies can be employed to mitigate this risk including in-line filtration, enhanced solvent selection and pulsed flow or backflushing. On the other hand, microreactors require different maintenance paradigms that are different from batch reactors that can be easily opened and cleaned between runs. However, industrial microreactor systems are typically constructed from multiple parallelized microchannel units. If clogging occurs in one module, it can be isolated, bypassed, and replaced online with a spare unit, ensuring continuous operation while maintenance is performed offline. Moreover, the use of corrosion-resistant materials like Hastelloy, Tantalum, or PTFE (as used in our study) is crucial for handling aggressive reagents like fuming nitric and concentrated sulfuric acids over extended periods, minimizing corrosion-related failures. By acknowledging and proactively designing solutions for these challenges, the transition from laboratory proof-of-concept to a robust industrial process becomes a manageable engineering task, further strengthening the case for the adoption of microchannel technology for hazardous reactions such as this one.
Conclusion
This study developed a microchannel reaction system for the oxidation and nitration of 3,5-dimethylpyridine. Through systematic optimization studies, optimal reaction parameters for microchannel reactor were established, achieving quantitative oxidation conversion of 100% yield within 4 h (vs 14 h of batch reactor synthesis) and a 67.3% isolated yield for the subsequent nitration step. Compared with conventional batch reactor synthesis, the microchannel reactor synthesis demonstrated substantial advancements such as reduced processing duration, enhanced productivity, minimized energy consumption and superior safety profile.
Supplemental Material
sj-docx-1-chl-10.1177_17475198251392346 – Supplemental material for Efficient production of 4-nitro-3,5-dimethylpyridine-N-oxide using microchannel technology
Supplemental material, sj-docx-1-chl-10.1177_17475198251392346 for Efficient production of 4-nitro-3,5-dimethylpyridine-N-oxide using microchannel technology by Wei-Si Li in Journal of Chemical Research
Footnotes
Acknowledgements
We acknowledge Southeast University and Nanjing Red Sun Pharmaceutical Research Institute Co. for supporting this study.
Ethical considerations
This paper does not contain any studies with human or animal participants.
Consent to participate
This paper has no human participants and informed consent is not required.
Consent for publication
Not applicable.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We thank Nanjing Red Sun Pharmaceutical Research Institute Co. for technical support. We also thank the Analysis and Testing Center of Southeast University for providing analytical instrumentation support.
Declaration of conflicting interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.