
Abstract
Resistomycin, a microbial secondary metabolite, exhibits promising anticancer potential, yet its mechanism of action remains poorly understood. Here, we combined computational and biological assays to investigate its activity against Topoisomerase II (Topo II). Molecular docking predicted strong binding within the Topo II–DNA complex (–20.76 kcal/mol), while 200 ns molecular dynamics simulations confirmed a stable intercalation mode. Energy decomposition revealed van der Waals interactions as the dominant stabilizing force, with persistent hydrophobic and π-stacking contacts to DNA bases. This non-polar binding profile distinguishes resistomycin from classical Topo II inhibitors. In line with these predictions, resistomycin inhibited Topo II activity with an IC₅₀ of 21.48 nM, surpassing doxorubicin (30.16 nM). Cell-based assays further showed selective cytotoxicity across multiple cancer cell lines (IC₅₀ = 45.68–72.30 µM), while mechanistic studies in HCT-116 cells demonstrated dose-dependent apoptosis and G2/M cell cycle arrest. Collectively, these findings identify resistomycin as a potent DNA-intercalating Topo II inhibitor with a unique binding signature, supporting its potential as a scaffold for developing next-generation anticancer agents.
Introduction
DNA topoisomerase II (Topo II) represents a critical therapeutic target in cancer treatment, serving as the molecular target for some of the most clinically successful anticancer agents, including doxorubicin and etoposide.1,2 These Topo II poisons function by stabilizing the enzyme-DNA cleavage complex, leading to irreversible DNA damage and subsequent cell death. 3 However, their clinical utility is significantly limited by dose-limiting toxicities, particularly cardiotoxicity, 4 genotoxicity 5 and the development of drug resistance.6,7 These limitations have driven the ongoing search for novel Topo II inhibitors with improved selectivity and safety profiles.
Natural products have historically served as an invaluable source of anticancer agents, with approximately 60% of currently used chemotherapeutics originating from natural sources. 8 Among these, angucycline antibiotics have attracted particular interest due to their unique chemical structures and diverse biological activities.9,10 Resistomycin, a member of this class originally isolated from Streptomyces resistomycificus, has demonstrated intriguing biological properties including antibacterial, antifungal, and anticancer activities.11,12 While its antimicrobial effects have been relatively well-characterized, the molecular basis of its anticancer activity remains poorly understood, particularly its potential interaction with Topo II.
Recent advances in computational structural biology provide powerful tools for elucidating the molecular mechanisms of drug-target interactions at atomic resolution.13,14 These in silico approaches, when combined with traditional biochemical and cellular assays, offer a comprehensive strategy for the mechanistic investigation of potential anticancer agents. 15 – 17 Our team has implemented this combined strategy across several therapeutic areas, including the development of SARS-CoV-2 antivirals, 18 – 20 targeting microbial virulence,21,22 development of anticancer agents, 23 – 26 and delivery optimization. 27 – 29
In this study, we employ an integrated computational and experimental approach to characterize resistomycin’s mechanism of action as a Topo II inhibitor. Through molecular docking and extensive MD simulations, we elucidate its unique binding mode within the Topo II–DNA complex. Complementary biochemical assays quantify its inhibitory potency against Topo II, while cell-based studies evaluate its cytotoxic effects and mechanism of action in cancer cells. Our findings reveal that resistomycin represents a novel class of DNA-intercalating Topo II inhibitor with distinct binding characteristics that may offer advantages over currently used agents. This work not only advances our understanding of resistomycin’s anticancer mechanism but also provides a foundation for developing improved Topo II-targeted therapies.
Results and discussions
Computational studies
Molecular docking
Molecular docking studies were performed to investigate the binding interactions of resistomycin and doxorubicin (a reference DNA intercalator) within the DNA–Topo II complex (PDB ID: 3QX3). The negative free energy values obtained for both compounds confirmed the spontaneity and stability of their binding. Doxorubicin exhibited a strong binding affinity of −27.47 kcal/mol, facilitated by its planar aromatic core, which formed four hydrophobic interactions with Gua13, Thy9, and Ade12. In addition, the methyl group of its sugar moiety contributed two hydrophobic interactions with Gua13 and Cyt8. The compound also established five hydrogen bonds with Lys456, Gua10, Ade12, and Thy9, further stabilizing its intercalation within the DNA–Topo II complex (Figure 1).
(a) 3D binding and (b) 2D binding of doxorubicin with DNA–Topo II complex.
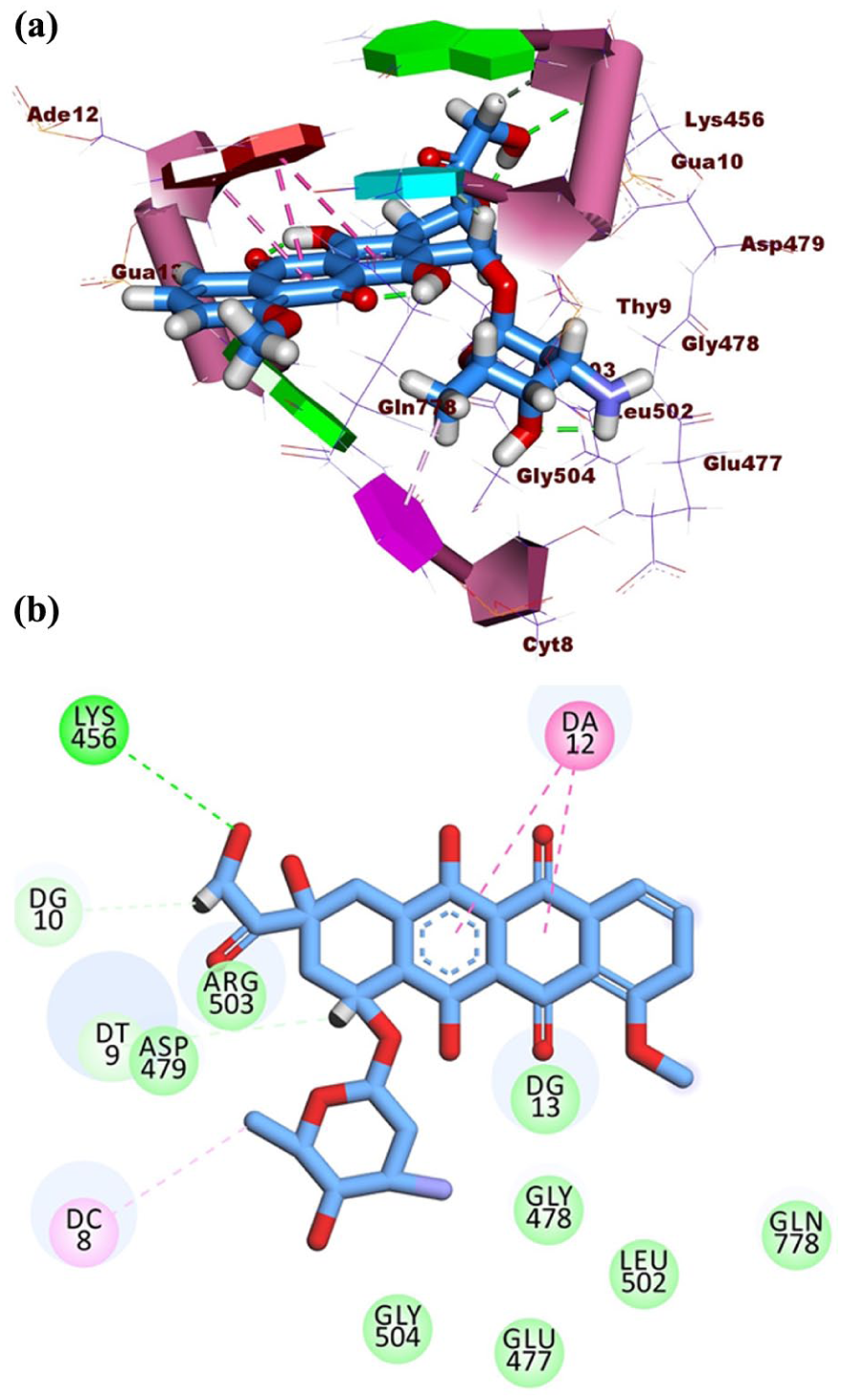
In comparison, resistomycin demonstrated a slightly lower but still significant binding affinity of −20.76 kcal/mol. Its planar aromatic system engaged in stacking interactions with the DNA base pairs Ade12, Gua13, and Thy9, while also forming seven hydrophobic contacts, including key interactions with Arg503 in the Topo II binding pocket. In addition, resistomycin formed three hydrogen bonds with Asp479, Thy9, and Gua13, contributing to its stable intercalation. Molecular dynamics simulations over 200 ns further confirmed the stability of resistomycin’s binding, with no significant dissociation observed (Figure 2).

(a) 2D binding and (b) 3D binding of resistomycin against DNA–Topo II.
Although doxorubicin displayed stronger binding affinity, resistomycin’s interaction profile suggests potential advantages in terms of selectivity and reduced off-target effects. Doxorubicin’s extensive hydrogen bonding and non-specific intercalation contribute to its high cytotoxicity but also to its severe side effects, such as cardiotoxicity and myelosuppression. In contrast, resistomycin’s more focused hydrophobic interactions with Arg503 and fewer hydrogen bonds may allow for selective Topo II inhibition while minimizing DNA damage in normal cells. The reduced number of hydrogen bonds relative to doxorubicin may also lower the risk of nonspecific DNA binding, potentially decreasing mutagenic risks associated with conventional intercalators.
Molecular dynamics simulation analysis
To further investigate the binding stability and interaction dynamics of resistomycin within the DNA–Topo II complex, a 200-ns MD simulation was performed. The protein maintained its structural integrity throughout the simulation, with a stable average root mean square deviation (RMSD) of approximately 3.7 Å (Figure 3(a)), indicating a globally stable protein conformation in the presence of resistomycin. In contrast, resistomycin exhibited distinct conformational flexibility over the course of the trajectory. Initially, the RMSD of the ligand remained stable at an average of ~4 Å during peaking at ~9 Å before returning to its original average toward the end of the simulation. This fluctuation reflects an adaptive binding behavior rather than ligand dissociation, as confirmed by center-of-mass distance analysis and visual inspection of superimposed ligand the first 60 ns, reflecting early binding equilibrium (Figure 3(b)). However, a transient increase in RMSD was observed between 60 and 120 ns, conformations at 0, 20, 100, and 170 ns (Figure 3(c)). These conformational adjustments suggest that resistomycin explores alternative binding orientations within the intercalation site to reach a more energetically favorable pose.
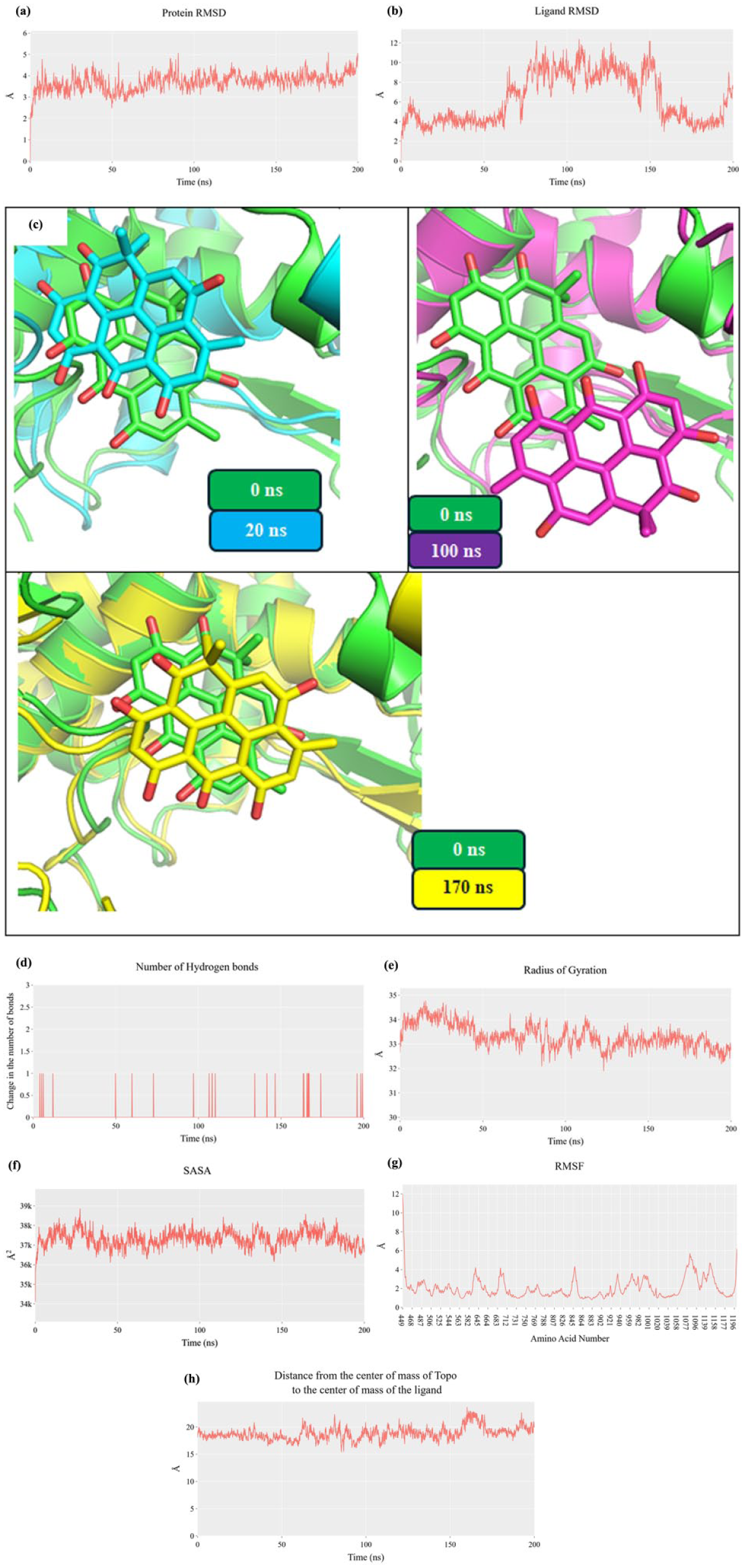
Molecular dynamics trajectories (200 ns) of resistomycin bound to DNA–Topo II complex. (a) Topo-II RMSD; (b) resistomycin RMSD; (c) Structural overlays at 0 ns (green sticks), 20 ns (cyan sticks), 100 ns (magenta sticks), 170 ns (yellow sticks); (d) hydrogen bond count; (e) RoG; (f) SASA; (g) Cα fluctuations; (h) distance from the center of mass of resistomycin and Topo II.
Hydrogen bond analysis showed that resistomycin formed relatively few polar interactions with the protein throughout the simulation, with a maximum of one hydrogen bond detected at any point (Figure 3(d)). This indicates that the ligand’s binding is driven predominantly by hydrophobic and π-stacking interactions rather than classical hydrogen bonding. Despite this, the center-of-mass distance between resistomycin and the DNA–Topo II complex remained consistent at approximately 18 Å, further supporting a stable binding mode. In addition, the radius of gyration (RoG) of the protein complex showed a brief elevation but subsequently stabilized around 33 Å (Figure 3(e)), while solvent accessible surface area (SASA) remained largely unchanged at ~37,500 Ų (Figure 3(f)), suggesting the system retained a compact and solvent-exposed architecture. Minor fluctuations in Cα atom positions were detected (Figure 3(g)), mainly within loop regions, which are expected to exhibit higher mobility.
MD simulations revealed that resistomycin undergoes noticeable conformational fluctuations within the Topo II–DNA complex, with RMSD values ranging between 4 and 9 Å. Despite this dynamic behavior, resistomycin persistent hydrophobic and π-stacking interactions with key DNA bases, supporting a stable intercalation mode. Such flexibility may reflect the ability of resistomycin to adapt its orientation within the DNA binding pocket, thereby sustaining favorable non-polar contacts even under conformational shifts of the complex. This dynamic adaptability is likely to contribute to the overall binding stability (ΔG = –27.63 kcal/mol, molecular mechanics/generalized born surface area (MM-GBSA)) and may underlie a distinct selectivity profile compared to classical Topo II inhibitors that rely more heavily on polar interactions. Taken together, these findings suggest that resistomycin’s structural flexibility supports a robust intercalation mechanism while potentially offering a unique binding signature that could be exploited in the design of optimized analogues.
MM-GBSA calculations
To quantify the thermodynamic favorability and driving forces behind the interaction of resistomycin with the DNA–Topo II complex, MM-GBSA calculations were conducted on representative frames extracted from the molecular dynamics trajectory. The analysis yielded an average total binding free energy of –27.63 kcal/mol, indicating a moderately strong and spontaneous interaction between resistomycin and the target complex (Figure 4). This binding energy falls within the range typically observed for small-molecule intercalators and suggests sufficient affinity to warrant further preclinical investigation.

MM-GBSA binding energy components of resistomycin in complex with DNA–Topo II.
A detailed breakdown of the energy components revealed that the majority of the favorable interaction energy stems from van der Waals (VDW) contributions, with a substantial energetic value of –44.3 kcal/mol. This dominant VDW component underscores the importance of hydrophobic and π–π stacking interactions in stabilizing the ligand within the binding site. These forces are especially relevant for resistomycin, which possesses a rigid, planar phenazine core—a structural feature known to promote efficient intercalation between DNA base pairs. The flat aromatic surface facilitates stacking interactions with nucleobases, maximizing VDW contact while minimizing steric hindrance.
In contrast, electrostatic interactions contributed only –15.41 kcal/mol to the total binding energy. This relatively modest value reflects the limited number of charged or polar functional groups on resistomycin available for hydrogen bonding or ionic interactions with the surrounding residues. The molecule’s primary hydrophobic character limits its capacity to engage in strong polar interactions, which is consistent with prior observations from both docking and MD hydrogen bond analysis, where few persistent polar contacts were observed.
Together, these findings suggest that hydrophobic stabilization, driven by aromatic stacking and non-polar contacts with the DNA helix, serves as the primary binding mechanism of resistomycin. This interaction profile aligns well with classical intercalators such as actinomycin D and certain anthracyclines, which also rely heavily on shape complementarity and hydrophobic insertion. The energetic data support the hypothesis that resistomycin’s binding mode is structurally optimized for intercalation, rather than groove binding or polar anchoring, which may offer distinct pharmacological advantages in specific therapeutic contexts, especially where resistance to traditional hydrogen bond-dependent drugs is observed.
Residue decomposition analysis (Figure 5) provided detailed insight into the atomic-level interactions that contribute to the binding affinity of resistomycin within the DNA–Topo II complex. The per-residue energy contributions revealed that the dominant binding interactions originate from the DNA component, rather than the protein, suggesting that resistomycin engages primarily with the nucleic acid substrate. Among the DNA residues analyzed, four nucleobases emerged as key energetic hotspots: Thymine 9 (–5.04 kcal/mol), Cytosine 8 (–4.07 kcal/mol), Adenine 12 (–2.7 kcal/mol), and Guanine 13 (–2.21 kcal/mol). The negative binding energy values associated with these bases indicate favorable and stabilizing interactions, each contributing substantially to the total free energy of the ligand–complex system.

Residue-based free energy decomposition of resistomycin–DNA–Topo II interactions.
The identity and positioning of these nucleobases are particularly significant. They are all situated within the central region of the DNA duplex adjacent to the intercalation site, and each has been previously implicated in π–π stacking, hydrophobic contact formation, and groove interactions in complexes with known DNA intercalators. The strongest contribution from Thymine 9 and Cytosine 8 suggests that these bases are likely serving as primary stacking surfaces for the planar phenazine core of resistomycin. This is consistent with the protein–ligand interaction fingerprint (ProLIF) interaction profile, which confirmed high-frequency stacking and hydrophobic interactions at these sites. The additional stabilizing interactions with Adenine 12 and Guanine 13 further reinforce the notion of a multibase intercalation footprint.
These energetic patterns, together with the spatial clustering of these nucleotides, support the conclusion that resistomycin binds deeply and selectively within the DNA double helix, favoring an intercalative mode of action rather than shallow minor-groove binding. The limited energetic contributions from protein residues further emphasize that the binding site selectivity is dictated more by DNA shape complementarity and electronic compatibility than by direct interactions with amino acid side chains from the topoisomerase II enzyme.
ProLIF analysis
To further characterize the nature and persistence of molecular interactions between resistomycin and the DNA–Topo II complex, a ProLIF analysis was performed across the 200 ns MD trajectory. This analysis revealed that resistomycin established consistent hydrophobic interactions with Cytosine 8 (chain E) and Thymine 9 (chain F), present in over 99% of the simulation frames (Figure 6). These high-frequency contacts indicate a strong and sustained association between the ligand’s aromatic core and these two DNA bases, further validating the energetic contributions identified in the residue decomposition analysis.
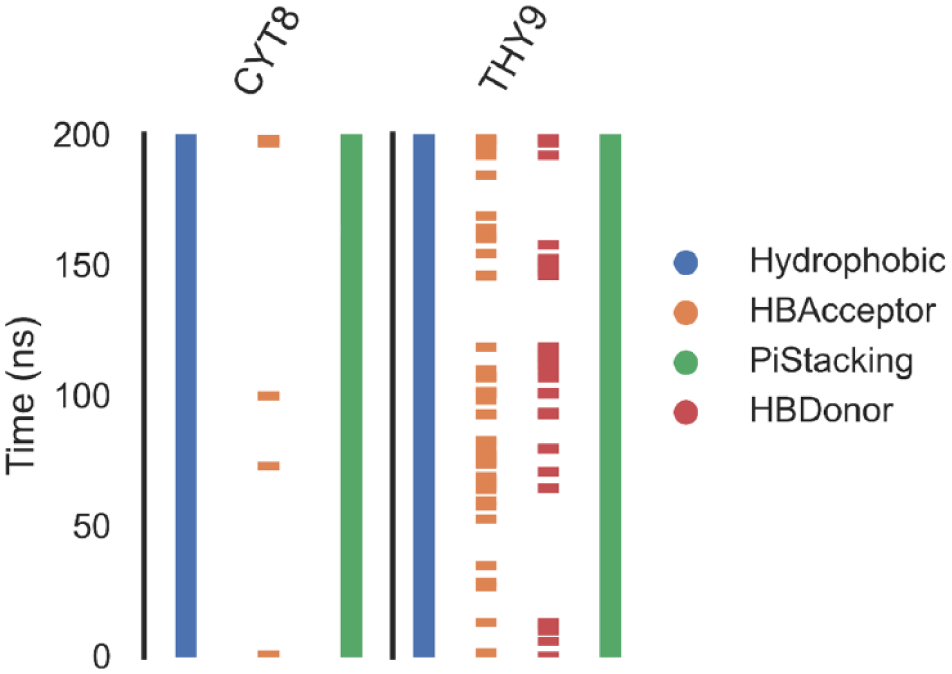
ProLIF fingerprint analysis of resistomycin interactions during MD simulation.
In addition to general hydrophobic interactions, ProLIF detected π-stacking interactions with exceptional frequency—specifically, 78.8% occurrence with Cytosine 8% and 82.6% with Thymine 9. These results underscore the importance of π–π interactions as a dominant stabilizing force in the resistomycin binding mechanism. The observed stacking behavior is highly characteristic of classical intercalative ligands, whose planar aromatic systems slide between DNA base pairs and engage in π-electron overlap to maximize VDW contact and base stacking. The persistence and specificity of these stacking events strongly suggest that resistomycin’s phenazine scaffold is structurally and electronically compatible with the DNA base pair environment.
Protein–ligand interaction profiler
To visualize these interactions in three dimensions, frames representing the most populated cluster of binding conformations were analyzed using the protein–ligand interaction profiler (PLIP). The PLIP output (Figure 7) clearly illustrated resistomycin intercalated between adjacent base pairs, close to Cytosine 8 and Thymine 9. The phenazine ring system was seen to adopt a planar orientation parallel to the base pair planes, reinforcing the stacking interactions observed in the fingerprint analysis. Notably, the ligand consistently maintained this orientation across three clusters, suggesting not only transient interaction but also geometric preference for intercalation as a primary binding mode.

3D Visualization of resistomycin binding pose using PLIP and PyMOL.
Taken together, these findings offer strong structural evidence that resistomycin binds through a classical intercalative mechanism, driven primarily by hydrophobic and π–π stacking interactions with key nucleobases. The high frequency and stability of these interactions throughout the trajectory reinforce the earlier MM-GBSA and residue decomposition results, collectively supporting the notion that resistomycin’s phenazine core provides a robust framework for selective DNA intercalation. These interaction characteristics highlight its potential as a lead structure for the development of topoisomerase II inhibitors that exploit DNA-mediated binding over enzyme-specific anchoring.
Overall, the MD simulation results suggest that resistomycin maintains a stable yet flexible binding mode within the DNA–Topo II complex, dominated by base-specific π-stacking and hydrophobic interactions. Despite the absence of extensive hydrogen bonding, the ligand remained associated throughout the simulation, showing adaptive intercalation behavior. These findings complement the docking analysis and reinforce resistomycin’s potential as a viable lead compound for Topo II inhibition and anticancer drug development.
An essential aspect worth considering is whether resistomycin’s unique DNA-centric and predominantly non-polar binding mode could offer advantages in overcoming resistance mechanisms commonly associated with classical Topo II inhibitors. Many clinically used agents, such as doxorubicin and etoposide, encounter reduced efficacy due to efflux by multidrug resistance transporters, altered drug–protein interactions, or mutations that weaken polar contacts within the Topo II–DNA cleavage complex. 30 – 32 In contrast, resistomycin’s reliance on hydrophobic and π-stacking interactions with DNA bases, rather than extensive polar contacts with the protein, may reduce susceptibility to such resistance pathways. 33 While this remains speculative and awaits direct experimental validation in resistant models, our computational analyses and in vitro potency raise the possibility that resistomycin may retain activity where conventional Topo II inhibitors are compromised. Future studies explicitly addressing resistance will be critical to determine whether this distinctive binding profile translates into therapeutic advantage.
Biochemical assays
Topo-II inhibition
Resistomycin demonstrated potent inhibition of Topo-II activity, exhibiting an IC50 value of 21.48 nM (Figure 8). This inhibitory potency surpassed that of the clinically used reference compound doxorubicin (IC50 = 30.16 nM), suggesting a potentially more effective mechanism of enzyme targeting. The 1.4-fold greater potency of resistomycin compared to doxorubicin indicates stronger binding affinity or more efficient interference with the Topo II catalytic cycle.
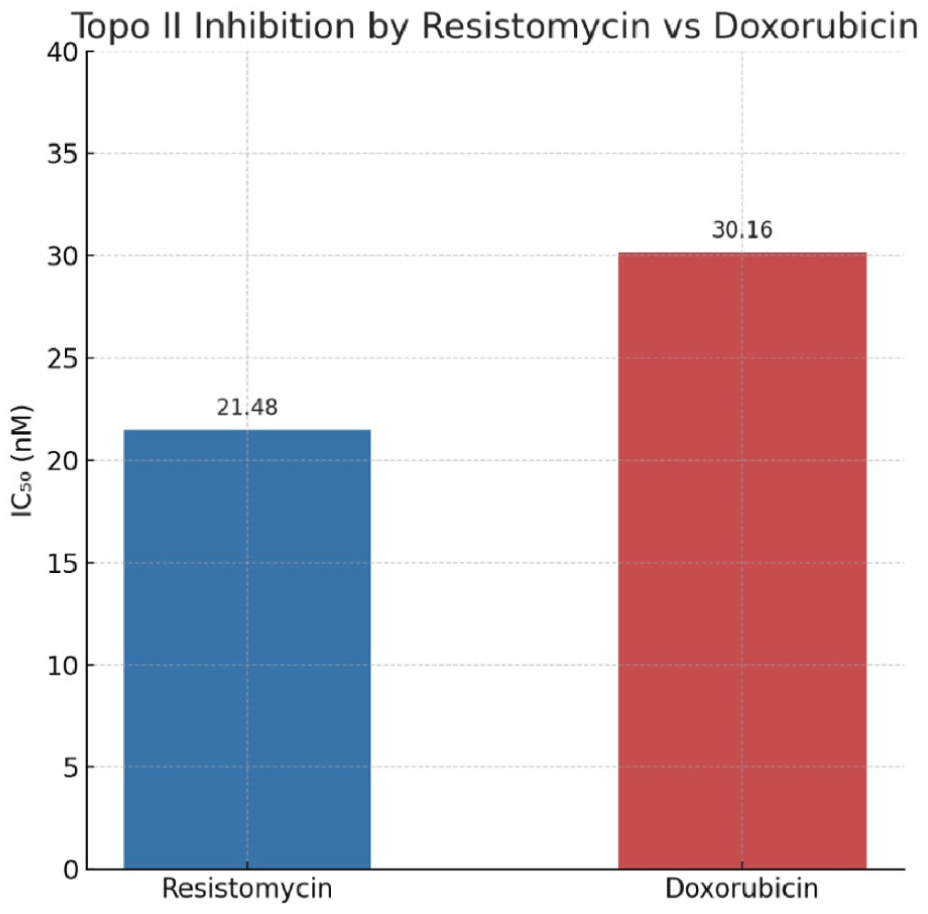
Topo II inhibition by resistomycin versus doxorubicin (IC₅₀).
While doxorubicin and other anthracyclines typically interact with both the protein and DNA components of the Topo II–DNA complex, computational analyses suggest resistomycin may preferentially target the DNA moiety. This DNA-centric mechanism of inhibition could account for its enhanced potency in the enzymatic assay while potentially offering advantages in terms of selectivity and reduced off-target effects.
Cytotoxicity
The cytotoxicity data comparing resistomycin and doxorubicin (DOX) across multiple cancer cell lines reveal several key advantages of resistomycin as a potential lead compound for anticancer drug development (Table 1 and Figure 9). While DOX exhibits superior potency, as expected from a clinically established chemotherapeutic, resistomycin demonstrates consistent mid-micromolar activity across diverse cancer types, suggesting a broad-spectrum effect that could be optimized for therapeutic use.
Cytotoxicity profiles of resistomycin and doxorubicin in normal and cancer cell lines (IC₅₀ in µM, mean ± SD, n = 3).

Statistical significance was determined using an unpaired t-test (p < 0.01) when comparing resistomycin and DOX treatments for each cell line relative to the control.

Comparative cytotoxicity of resistomycin and doxorubicin across several cancer cell lines.
One of the most compelling advantages of resistomycin is its moderate yet selective cytotoxicity, particularly in certain cell lines such as HCT-116 (colorectal carcinoma, IC50 = 45.68 ± 2.7 µM) and MDA-231 (triple-negative breast cancer, IC50 = 49.78 ± 2.7 µM). Unlike DOX, which shows significant variability in efficacy (e.g. 4-fold difference between MDA-231 and Caco-2), resistomycin’s activity varies only ~1.6-fold across the tested cell lines. This suggests a more balanced mechanism of action, potentially targeting conserved pathways in cancer cells rather than relying on highly specific interactions that may lead to resistance.
In addition, resistomycin’s higher IC50 values compared to DOX could be advantageous from a toxicity perspective. DOX’s extreme potency is often accompanied by severe side effects, including cardiotoxicity, which limits its clinical utility. resistomycin’s milder cytotoxicity profile may indicate a better therapeutic window, reducing off-target effects while still maintaining anticancer activity. This makes it an attractive candidate for structural modification—chemical optimization could enhance its potency while preserving its potentially safer profile.
Another promising aspect is resistomycin’s performance in DOX-resistant contexts. For instance, in Caco-2 (colorectal adenocarcinoma), which shows relative resistance to DOX (IC50 = 12.49 ± 1.1 µM), resistomycin still exhibits measurable activity (IC50 = 72.30 ± 3.8 µM). This raises the possibility that resistomycin operates through a distinct mechanism. Further mechanistic studies could clarify whether resistomycin could be used as an alternative or adjunct therapy in DOX-resistant cancers.
The observed discrepancy between the strong enzymatic inhibition of Topo II by resistomycin and its comparatively weaker cytotoxicity in cellular assays is noteworthy. This difference may be attributed to several factors, including limited cellular uptake, restricted membrane permeability, active efflux by transport proteins, or metabolic instability within the cellular environment. Such phenomena are frequently encountered with natural products and structurally complex molecules, where high target affinity does not always translate into equally potent cellular effects. Although dedicated experiments to assess resistomycin’s pharmacokinetics, stability, and uptake were beyond the scope of the present study, these aspects represent important directions for future investigation. Moreover, the mid-micromolar cytotoxicity observed in multiple cancer cell lines still indicates a selective anticancer effect, suggesting that optimization strategies such as structural modification or advanced delivery systems may enhance its bioavailability and therapeutic efficacy.
Cell cycle
Resistomycin treatment induced significant perturbations in the cell cycle distribution of HCT-116 colon cancer cells, demonstrating its potent antiproliferative activity (Table 2 and Figure 10). The most pronounced effect was observed in G2/M phase, where the cell population decreased by 38% from 44.40% in control cells to 27.40% following treatment, indicating a partial but substantial blockade of mitotic progression. This G2/M arrest was accompanied by an even more dramatic 46% reduction in S-phase cells (from 10.25% to 5.55%), revealing robust inhibition of DNA replication. G1 phase was less affected, with only a 13% reduction (17.75% to 15.40%), implying that resistomycin’s primary actions target later cell cycle stages.
Cell cycle arrest induced by resistomycin in HCT-116.


Effect of resistomycin on cell cycle progression in HCT-116 cells.
These cell cycle effects provide mechanistic insights into resistomycin’s anticancer activity. The dual-phase impact on both S-phase and G2/M populations suggests a complex mechanism involving direct interference with DNA replication machinery followed by checkpoint-mediated arrest before mitotic entry. The S-phase suppression is particularly noteworthy and consistent with Topo II’s critical role in DNA replication, potentially reflecting either direct enzyme inhibition or DNA intercalation effects. The partial G2/M arrest may result from activation of DNA damage checkpoints due to unresolved topological stress or incomplete chromosome decatenation. The relatively modest increase in apoptotic cells indicates that cell cycle disruption precedes and likely triggers subsequent programmed cell death, rather than resistomycin acting as a direct apoptosis inducer.
Such broad-spectrum cell cycle activity could potentially reduce the likelihood of resistance development and create opportunities for synergistic combinations with other cell cycle-targeted therapies.
Apoptosis induction
Resistomycin treatment induced profound changes in cell death pathways in HCT-116 colon cancer cells, demonstrating its potent cytotoxic effects (Table 3 and Figure 11). The proportion of viable cells dramatically decreased from 98.46% in untreated controls to just 4.41% following resistomycin exposure, indicating near-complete loss of cell viability. This massive reduction in viable cells was accompanied by substantial increases in all forms of cell death, with apoptosis emerging as the predominant mechanism. Early apoptotic cells rose from a minimal 0.10% in controls to 28.81% after treatment, representing a remarkable 288-fold increase, while late apoptotic cells showed a 31-fold increase from 1.12% to 34.69%. Together, these apoptotic populations accounted for 63.5% of all cells, demonstrating resistomycin’s exceptional ability to activate programmed cell death pathways.
Viability and death pathways in HCT-116 following resistomycin exposure.


Apoptosis induction by resistomycin in HCT-116 cells.
The cell death profile revealed that resistomycin triggers a complex pattern of cytotoxicity involving both apoptotic and necrotic mechanisms. While apoptosis clearly dominated, evidenced by the combined 63.5% of cells in early and late apoptosis, necrosis also showed a substantial 100-fold increase from 0.32% to 32.10%. This necrotic population likely represents multiple phenomena: true primary necrosis in some cells, secondary necrosis in apoptotic cells that completed the death program, and possibly some necroptosis. The 2:1 ratio of apoptotic to necrotic cells suggests that apoptosis is the primary death mechanism, with necrosis occurring either as a consequence of overwhelming cellular damage or as a secondary event following apoptosis. This mixed death profile is particularly interesting therapeutically, as it combines the controlled, immunologically “quiet” nature of apoptosis with the potentially immunostimulatory effects of necrosis, which may help trigger antitumor immune responses.
These findings provide crucial mechanistic insights that complement the observed cell cycle effects and cytotoxicity data. The massive induction of apoptosis explains the potent anticancer activity reflected in the IC50 value of 45.68 μM, while the substantial necrotic component may contribute to additional therapeutic benefits through immune system activation. The cell death patterns are consistent with resistomycin’s identified mechanism as a Topo II inhibitor, as such agents typically induce DNA damage that triggers apoptotic pathways. However, the unusually high percentage of cells undergoing late apoptosis and necrosis suggests that resistomycin may have additional mechanisms that ensure completion of the cell death program, rather than allowing temporary cell cycle arrest and potential recovery. This comprehensive induction of cell death supports resistomycin’s potential as an effective anticancer agent and warrants further investigation into its precise molecular targets and death pathway activation mechanisms.
Interestingly, our findings are consistent with previously published reports showing that Resistomycin exhibits significant anti-cancer activity through multiple mechanisms. At the cellular level, resistomycin inhibits proliferation and induces apoptosis by upregulating pro-apoptotic markers (Bax, cleaved caspase-3, cytochrome c) and downregulating anti-apoptotic Bcl-2 in colorectal, prostate, and liver cancer models. In addition, resistomycin causes cell cycle arrest, particularly at the G2/M phase, thereby halting uncontrolled proliferation in hepatocellular carcinoma and prostate cancer cells. 34 – 36
Beyond in vitro evidence, resistomycin has also demonstrated promising in vivo efficacy. In mouse models of human hepatocellular carcinoma, treatment with resistomycin significantly reduced tumor growth, consistent with activation of the p38 MAPK pathway, apoptosis, and G2/M arrest in tumor tissues. Notably, this effect was achieved with minimal toxicity to normal tissues, indicating a favorable therapeutic index. 36 Moreover, studies in oral cancer models using a water-soluble derivative of resistomycin (4-dmH) showed enhanced tumor volume reduction and greater downregulation of cancer-associated proteins (tNOX and SIRT1) compared to control. 37
Although molecular docking, MD simulations, and MM-GBSA analyses are widely recognized as reliable for exploring ligand–target interactions and generating mechanistic hypotheses, these computational tools inevitably rely on certain assumptions such as predefined force fields, simplified solvation models, and finite simulation timescales. In this work, the strong agreement between docking scores, dynamic stability over 200 ns, and energy decomposition profiles adds confidence to the robustness of the predicted intercalation mode of resistomycin. While such models cannot fully reproduce the complexity of the cellular microenvironment, they effectively capture the dominant binding features, such as hydrophobic and π-stacking interactions with DNA bases. Importantly, these in silico findings were not considered in isolation but were reinforced by biochemical Topo II inhibition assays and cellular experiments demonstrating apoptosis and G2/M arrest. Together, this integrated strategy minimizes the impact of computational assumptions and ensures that the conclusions drawn are both credible and biologically meaningful.
In addition, it is important to note that the clinical application and pharmacokinetic properties of resistomycin have not yet been reported, and further studies are required to establish its translational viability. Natural products frequently face challenges such as poor aqueous solubility, rapid metabolic clearance, and limited systemic exposure, all of which may restrict therapeutic efficacy. Improving bioavailability and biodistribution will be essential to ensure adequate tumor penetration while reducing off-target effects. Another critical factor is safety: although resistomycin demonstrated selective cytotoxicity in vitro, its toxicity profile in normal tissues remains undefined and will require careful evaluation in preclinical models. To address these limitations, multiple strategies could be considered, including solubility enhancement (e.g. salt formation, prodrug design), stabilization against metabolic degradation through structural modification, and advanced delivery approaches such as liposomal encapsulation, polymeric nanoparticles, or antibody–drug conjugates. These approaches may improve stability, selectivity, and overall pharmacological performance. Ultimately, systematic in vivo pharmacokinetic, biodistribution, and toxicity studies—followed by translational assessments in animal models—will be essential to define parameters such as maximum tolerated dose, therapeutic index, and antitumor efficacy. Collectively, these considerations will be pivotal in advancing resistomycin toward clinical application and guiding rational strategies for its future development as a potential anticancer agent.
Experimental
Molecular docking analysis
The binding interactions between resistomycin and the DNA–Topo II complex (PDB ID: 3QX3) were examined following our previously established protocol. 38 The analysis included (1) protein preparation (hydrogen addition, protonation state optimization, and energy minimization), (2) ligand preparation (conformational sampling and charge assignment), and (3) docking grid placement at the ATP-binding site of Topo II. Detailed procedures are provided in Supplementary Information (Section S1).
MD simulation studies
A 200-ns all-atom MD simulation of the resistomycin–Topo II complex was performed using GROMACS 2021 with the CHARMM36 force field. The system was solvated in a TIP3P water box (10 Å buffer) with 150 mM NaCl, followed by energy minimization (steepest descent), equilibration (NVT and NPT ensembles), and production runs (2 fs timestep). Comparative trajectory analysis between apo (ligand-free) and holo (resistomycin-bound) Topo II revealed critical structural changes. 39 Full simulation parameters and CHARMM-GUI 40 input configurations are documented in Supplementary Information Section S2.
Binding free energy calculations (MM-GBSA)
Binding energetics were computed using the gmx_MMPBSA toolkit with the MM-GBSA method. From 100 equally spaced trajectory frames, we determined: (1) total binding free energy (ΔG bind), (2) energy contributions (VDW, electrostatic, polar/nonpolar solvation), and (3) per-residue decomposition for residues within 10 Å of resistomycin. 41 The complete protocol is available in Supplementary Information (Section S3).
ProLIF analysis
Residue-specific interactions were analyzed across all simulation frames using ProLIF (v1.0.0). 42 Key interactions included the following:
Hydrogen bonds (distance <3.5 Å, angle >120°);
Hydrophobic contacts (⩽4.5 Å);
π-stacking/ionic interactions;
Interactions with >70% occurrence were considered biologically significant. Full interaction matrices are in Supplementary Dataset S4.
PLIP analysis
MD trajectory was processed as follows: 43 Clustering: TTClust algorithm (RMSD cutoff = 2.0 Å) identified three dominant conformations.
Frame Selection: Centroid structures from each cluster were extracted.
Mapping: PLIP webserver generated: • 2D interaction diagrams (hydrogen bonds, π-effects, salt bridges) • 3D visualizations (.pse files) rendered in PyMOL (v2.5.2).
4. Interaction persistence (%) across clusters was calculated.
5. PLIP files and PyMOL sessions are archived in the Supplementary Materials (Section S5).
Compound sourcing
Resistomycin was acquired from in-house stocks following prior isolation from Streptomyces sp. SP9. 35
In vitro Topo II inhibition assay
Resistomycin’s inhibitory activity was measured using a human Topo II ELISA kit. Full experimental conditions and image analysis methods are in the Supplementary Information.
Cytotoxicity and antiproliferative profiling
Cytotoxicity was assessed via MTT assay 44 across a panel of seven cancer cell lines:
HepG-2 (liver carcinoma);
MCF-7 and MDA-MB-231 (breast adenocarcinoma);
HCT-116 and Caco-2 (colorectal carcinoma);
PC-3 (prostate cancer);
HeLa (cervical adenocarcinoma).
Doxorubicin served as the positive control. Data are presented as mean ± SD from three independent replicates (Supplementary Information, Section S7).
Apoptosis and cell cycle analysis by flow cytometry
Mechanistic effects on HCT-116 cells were evaluated according to the previous reports 45 using:
Cycle distribution: Propidium iodide (PI) DNA staining.
Detection: Annexin V-FITC/PI dual staining.
Standard gating distinguished: • Cell cycle phases (G0/G1, S, G2/M); • Apoptotic populations (viable, early/late apoptotic, necrotic cells) Full protocols are in Supplementary Materials (Section S8).
Conclusion
This study provides compelling evidence that resistomycin functions as a potent and mechanistically distinct Topoisomerase II inhibitor with promising anticancer properties. Our integrated computational and experimental approach revealed that resistomycin preferentially targets the DNA component of the Topo II–DNA complex through strong intercalative interactions, as demonstrated by its stable binding energy (ΔG = −27.63 kcal/mol) and dominant hydrophobic/π-stacking contacts with Thy9 and Cyt8 bases. This DNA-centric binding mode, characterized by minimal protein interactions (⩽1 hydrogen bond), represents a significant departure from classical Topo II inhibitors like doxorubicin and may underlie resistomycin’s superior enzymatic inhibition (IC₅₀ = 21.48 nM vs doxorubicin’s 30.16 nM). The biological relevance of this unique binding mechanism was confirmed through comprehensive cell-based assays. Resistomycin exhibited selective cytotoxicity across multiple cancer cell lines while inducing hallmark effects of Topo II poisoning in HCT-116 cells, including substantial apoptosis (63.5% combined early/late) and G2/M phase arrest (27.4% vs 43.8% control). These cellular responses correlate well with the DNA intercalation activity observed in silico and suggest that resistomycin’s anticancer effects primarily stem from its ability to disrupt Topo II-DNA dynamics rather than direct protein inhibition. The combination of potent Topo II inhibition, selective cytotoxicity, and this novel DNA-targeted mechanism positions resistomycin as a promising scaffold for next-generation anticancer development. Its non-classical binding profile may offer several therapeutic advantages, including reduced likelihood of resistance development and potentially improved safety compared to current Topo II inhibitors. Future research should focus on structural optimization to enhance cellular uptake and in vivo validation of these promising properties, with the goal of developing more effective and selective Topo II-targeted therapies for clinical use.
Supplemental Material
sj-docx-1-chl-10.1177_17475198251385678 – Supplemental material for Resistomycin as a DNA-targeted topoisomerase II inhibitor: Computational and mechanistic insights into its anticancer potential
Supplemental material, sj-docx-1-chl-10.1177_17475198251385678 for Resistomycin as a DNA-targeted topoisomerase II inhibitor: Computational and mechanistic insights into its anticancer potential by Mohamed S Abdelfattah, Ibrahim H Eissa, Ahmad E Mostafa, Eslam B Elkaeed, Aisha A Alsfouk, Abdulla A Mahmoud, Ibrahim M Ibrahim and Ahmed M Metwaly in Journal of Chemical Research
Footnotes
Acknowledgements
The authors would like to thank AlMaarefa University, Riyadh, Saudi Arabia, for supporting this research.
Ethical Considerations
This study did not involve human participants, animal experimentation, or clinical data collection. As such, ethical review and approval by an Institutional Review Board were not required.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R116), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
All data generated or analyzed in this study are included in:
• The main manuscript
• Supplementary materials
Informed consent statement
Not applicable. The research was conducted exclusively using computational modeling and in vitro assays with commercially available cell lines, which do not require informed consent.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.