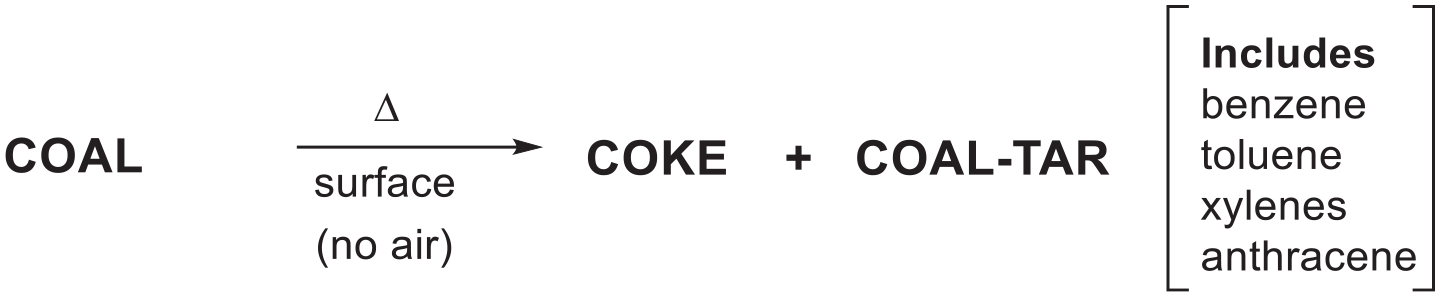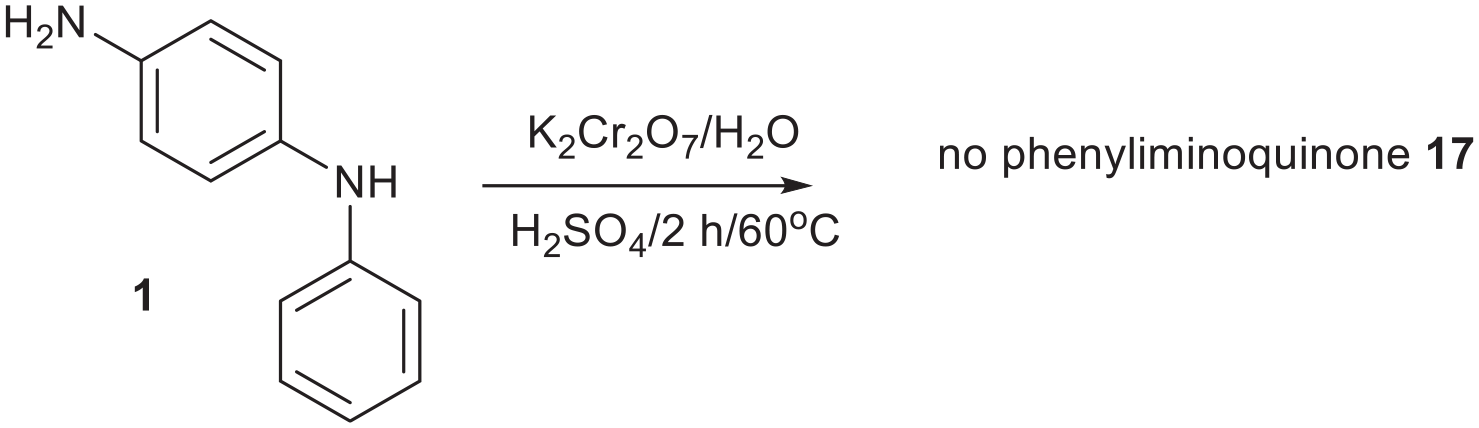A mixed ligand ZnCl2 complex of N-tolyl-p-phenylenediamine, ZnCl2(C13H14N2)2, was unexpectedly formed in situ in the Zn/EtOH/H+ reduction of 4-nitrophenylaminotoluene and was characterised by an X-ray single crystal structure determination. The Zn2+ cations were presumably formed by oxidation with the cHCl in EtOH used to clean the zinc metal surface. Two failed mauveine syntheses with preformed building blocks suggest that linear trimers are mauveine precursors in the William Henry Perkin 1856 mauveine synthesis. An unusual synthesis of phenyliminoquinone (C13H13N3O2) is reported which provides further evidence on the mechanism of mauveine formation.
Key intermediates from coal tar in the 19th century, for aniline dye synthesis.
Isolated from coal by Hofmanns student, Charles Blachford Mansfield.
Introduction
William Henry Perkin’s (WHP) patent of 1856 reported the synthesis of a purple dye, named mauveine, formed by the oxidation of aniline sulphate with potassium dichromate in water.1 Four years later, in 1860, Heinrich Caro prepared mauveine by a less practical method using a weak oxidant copper(II) chloride in water instead of potassium dichromate.2 The synthesis of aniline was possible on a large scale because benzene, containing about 10% toluene, was distilled from coal tar, a by-product from the conversion of coal to coke in the illuminating gas industry (Figures 1–3).3–6Figure 1 shows a piece of coal, which coal tar can be made from and a mauveine dyed silk lady’s day dress. An early review appears in ProQuest, From Coal to Colour: Once a Week, 1871; 8: 194–197. Aug 26th.7
Mauveine dye made from coal used for dyeing an 1860s silk lady’s day dress.
The formation of coal tar and some of its products.
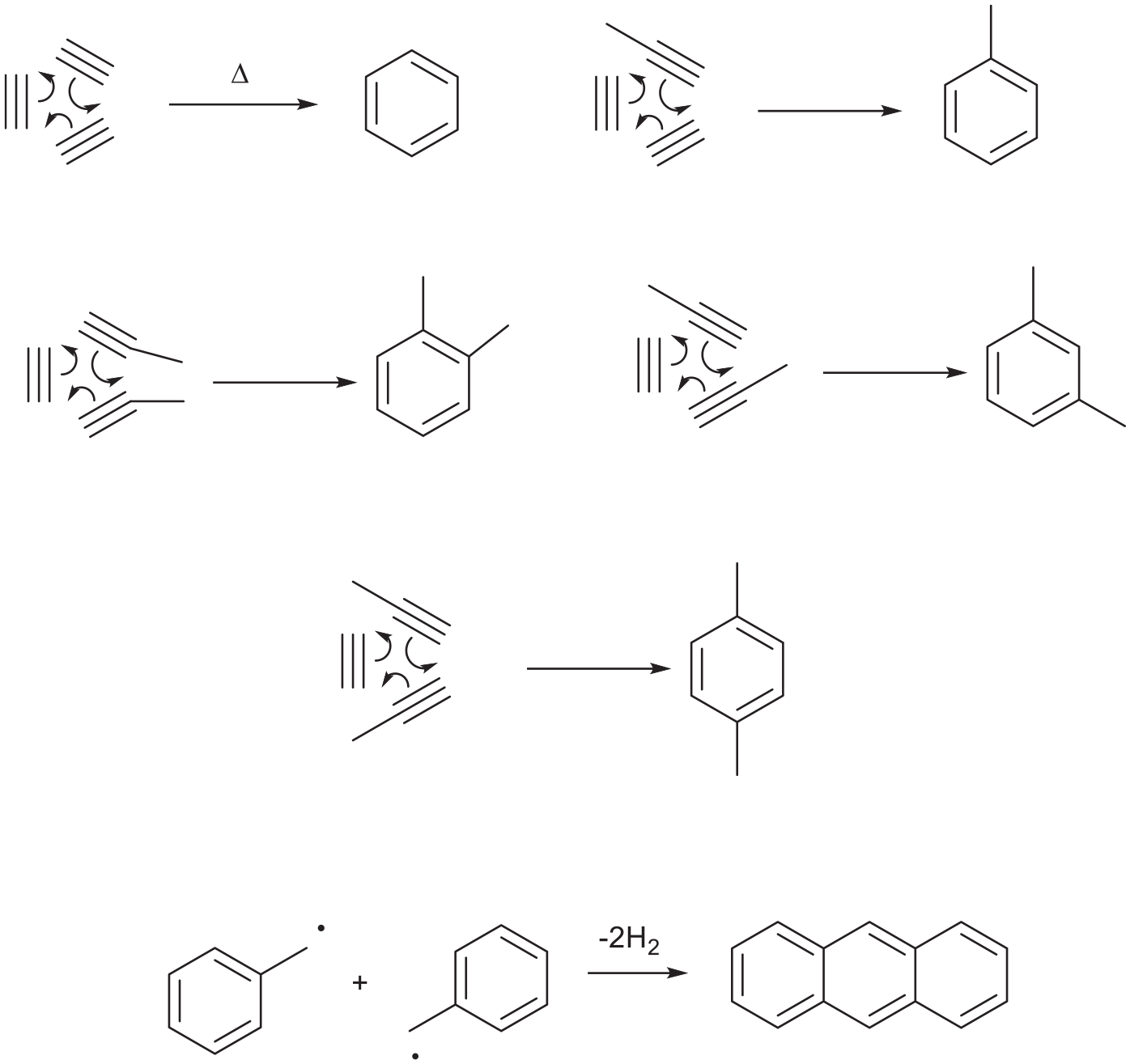
Proposed mechanism for forming key components of coal tar from acetylene and propyne.
Variations on the WHP method of mauveine synthesis have been reported,8–12 and some methods use preformed building blocks or early reactions from oxidising aromatic amines.13–20
Figure 2 shows some of the products that were distilled from coal tar. These include benzene, toluene, xylenes and anthracene. Figure 3 shows a proposed mechanism for the formation of these compounds. The cyclotrimerisation of acetylene on a hot surface could form benzene and cyclotrimerisations of acetylene with propyne could give toluene and xylenes.21 Anthracene from coal tar, a precursor to anthraquinone,22,23 could form via the dimerisation of two benzyl radicals. This thermal chemistry was pivotal to the coal tar or aniline dye industry. The benzene/toluene (90:10) mixture was nitrated then reduced to aniline/toluidine with iron in HOAc (Bechamp process).24
This thermal, gaseous, molecular chemistry without air, to make benzene and toluene4 was pivotal to the aniline dye industry and anthracene was used for making the anthraquinone dye alizarin.22,23
Discussion
N-Phenyl-p-phenylenediamine 1 is a useful preformed building block for making mauveine.25
An attempt was made to reduce the related methylated building block 4,26 but instead of the expected product, we obtained the in situ formed ZnCl2 coordination complex 5. The Zn2+ ions must come from the oxidation of zinc with cHCl/EtOH, which was added to clean the zinc metal surface. The spontaneous formation suggests compound 5 might be a useful catalyst. A different building block was then pursued.
Compound 5 (Figure 4) crystallises with one molecule in the asymmetric unit (Figure 5) in the monoclinic space group Ia. The zinc cation is coordinated by two chloride ions and two N-bonded neutral C13H14N2 ligands. As expected, the Zn–Cl bonds [2.240 (2) and 2.245 (2) Å] are significantly longer than the Zn–N bonds [2.041 (8) and 2.059 (8) Å]. In the N1-ligand, the dihedral angle between the phenyl rings is 53.3 (3)° with a corresponding value of 42.3 (3)° for the N3 ligand. The dihedral angle between the rings bonded to the zinc ion is 85.5 (2)°. In the extended structure of compound 5, the molecules are linked by N–H. . .Cl hydrogen bonds to generate (001) sheets.
A serendipitous synthesis of a Zn(II)Cl2 complex 5.
The molecular structure of compound 5 showing 50% displacement ellipsoids.
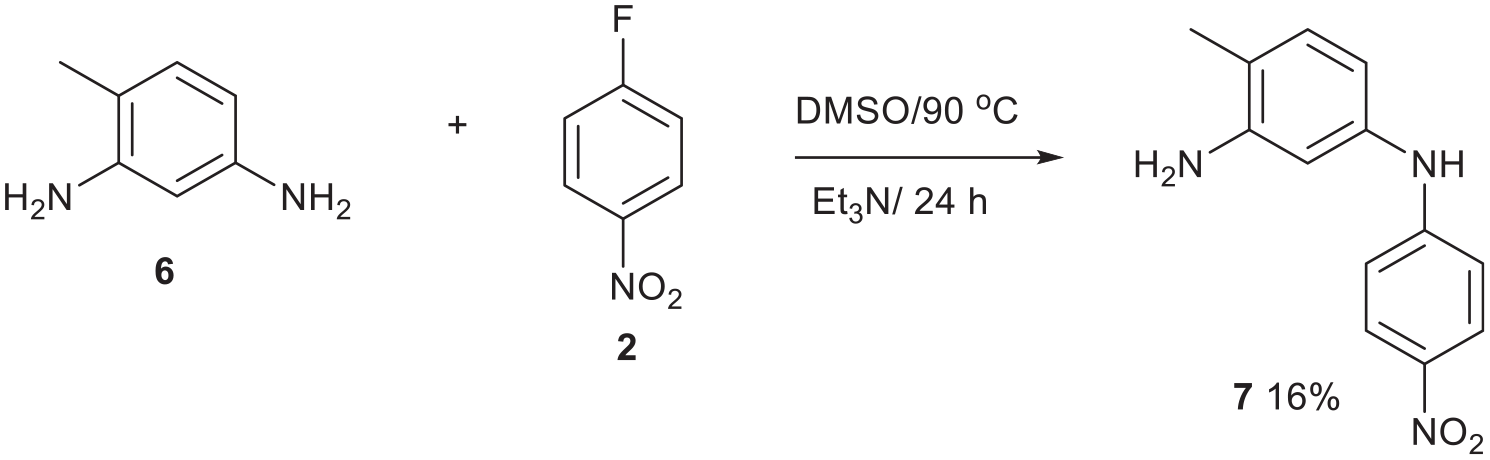
Compound 7 was prepared by a selective nucleophilic displacement of fluorine from 4-nitrofluorobenzene 2 (Figure 6). The primary amine ortho to the methyl group is more hindered and indeed it failed to react. The nuclear magnetic resonance (NMR) data were satisfactory, but arguably, they could have corresponded to the other isomer, so the structure was proved by an X-ray single crystal structure determination (Figure 7). Compound 7 is a preformed mauveine building block. The nitro group was not expected to hinder its reactivity because it would be involved in a favourable intramolecular cyclisation reaction to form a mauveine (see Figure 8).
Synthesis of compound 727,28 by a reaction of the less hindered primary aromatic amine.
The molecular structure of compound 7 showing 50% displacement ellipsoids.
First failed mauveine 8 synthesis.
The molecular structure of compound 7, which crystallises with one molecule in the asymmetric unit in the orthorhombic space group Pbca, is shown in Figure 7. The dihedral angle between the phenyl rings is 49.27 (3)° and the nitro group is almost coplanar with its attached ring [dihedral angle = 5.26 (13)°]. The C7–N1 bond [1.3660 (11) Å] is shorter than C4 – N1 [1.4085 (11) Å] presumably due to some conjugation of the N1 lone pair with the nitro group. In the extended structure of 7, N–H. . .O and N–H. . .N hydrogen bonds connect the molecules into a three-dimensional network.
Surprisingly, we isolated no mauveine from this reaction by working the reaction up19 and analysing a MeOH solution by TLC29,30 (Figure 8). The test for mauveine like this is quite sensitive.
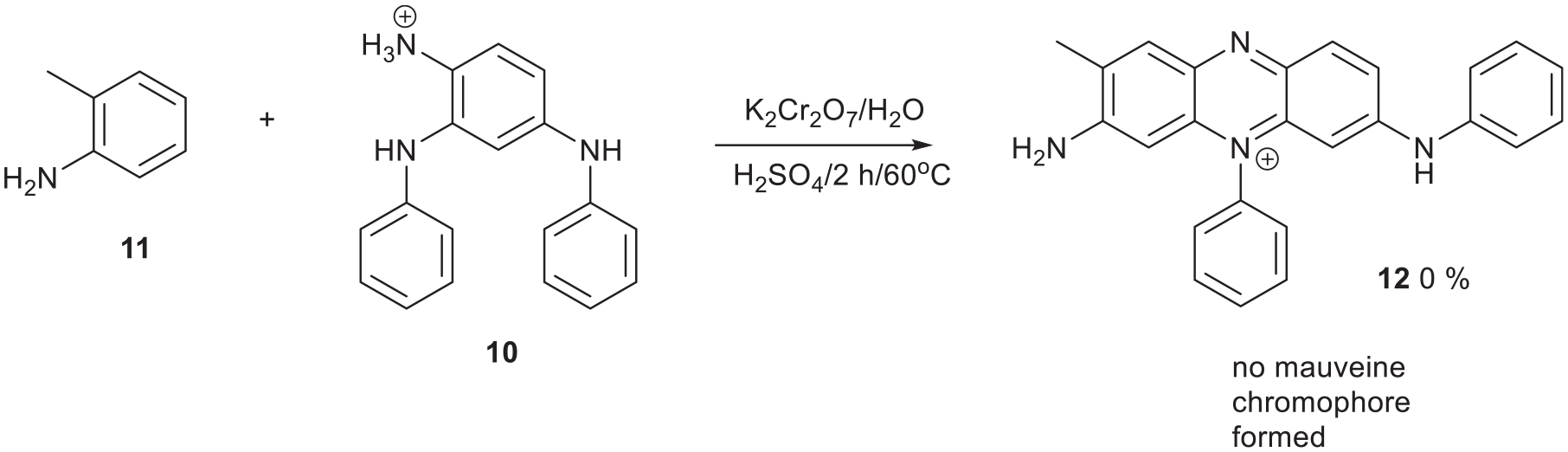
A second mauveine synthesis was attempted using the in situ–generated crude precursor 10 (Figure 9).31,32 [Compound 9 was previously prepared from 2,4-difluoronitrobenzene and aniline.19] This method of synthesis, using the HCl salt 10, gave the best 1H NMR data for it.
Precursor 10 synthesis.
Both these reactions failed to produce mauveine presumably because of subtle steric effects around the components (Figures 8 and 10). The N-aryl-p-phenylenediamine is expected to oxidise to an N-phenyl-p-phenylene imine, which initiates the reaction.
Second failed mauveine 12 synthesis.
The trimer33 synthesis of mauveine was reported previously using K3Fe(CN)6 as oxidant under basic conditions.33 The reaction does indeed work under the WHP conditions of mild acidity with K2Cr2O7.1 (Figure 11).
Synthesis of pseudomauveine 15 from a linear trimer 14 with potassium dichromate/H2O/H2SO4.
An attempt was made to make a linear trimer, like compound 14, by oxidising N-phenyl-para-phenylenediamine 1 in the presence of either 2,6-dimethylaniline 16 or N,N-dimethylaniline 18 (Figure 12 and 13). The proposed trimers are compounds 19 and 20. Trimers are believed to form as key intermediates in mauveine synthesis.33,34 No linear trimer was isolated but phenyliminoquinone 17 forms as a stable blue compound (Figure 12).35 It is a strikingly coloured reaction, and the product elutes with MeOH on silica whereas mauveine is more polar requiring aqNH3/MeOH (20:80). The reaction to form phenyliminoquinone 17 failed when compounds 16 or 18 were absent (Figure 14, right spot). No trimers have been isolated in mauveine syntheses, but this reaction suggests that here a trimer is forming but it is hydrolytically unstable. If this is applied to mauveine synthesis, it suggests a reason as to why no trimers have been observed: they are too unstable and must be intercepted quickly. The failure of the reactions in Figure 12 suggests that linear trimers are the short-lived intermediates that lead to mauveine.33
Syntheses of phenyliminoquinone 17 using N-phenyl-p-phenylenediamine 1 with a coupling amine.
Postulated short-lived linear trimer precursors for compound 17.
Left: Blue spot of phenyliminoquinone 17 after a column from reacting 2,6-dimethylaniline 16 or N,N-dimethylaniline 18 with N-phenyl-p-phenylenediamine 1 and K2Cr2O7 in dilute sulfuric acid. The failed reaction on the right is from Figure 15.
Failed synthesis of phenyliminoquinone 17 from N-phenyl-p-phenylenediamine 1.
Conclusion
This work suggests that linear trimers are likely to be involved in mauveine synthesis but these trimers are hydrolytically unstable which can explain why they have not been observed in these syntheses. In a situation where trimers are not intercepted, using compounds 16 and 18 as intercepting reagents, then a stable, blue hydrolysis product phenyliminoquinone 17 is formed. This does not form unless compounds 16 or 18 are present to intercept an oxidised form of N-phenyl-p-phenylenediamine leading to a hydrolytically unstable oxidised trimer that gives 17. Oxidised linear trimers formed in mauveine syntheses are also likely to be hydrolytically unstable and should be intercepted quickly by an aromatic amine. Two failed mauveine syntheses suggest that linear trimers are precursors to mauveine in the WHP mauveine synthesis. The zinc dust reduction of compound 4 gave an unexpected metal complex using up the ZnCl2, which formed in situ by the acid cleaning of the zinc oxide surface. It is suggested that the zinc is best cleaned with cHCl/EtOH then the solvent may be changed for fresh EtOH/H+. The two primary amines of 2,4-diaminotoluene 6 can be distinguished in their reactivity owing to their different steric hindrance.
4-Nitrofluorobenzene 2 (1.0 g, 7.1 mmol) and p-toluidine 3 (1.52 g, 14.2 mmol) were mixed in EtOH (5 mL) and heated in a pressure vessel at 150 °C for 24 h. After cooling the reaction mixture was diluted with water (200 mL), extracted with DCM (100 mL), backwashed with water (100 mL) and dried over MgSO4. The product was purified on a silica column. Elution with DCM/light petroleum ether (25:75) followed by DCM/light petroleum ether (50:50) gave the title compound (430 mg, 27 %) as an orange solid with spectroscopic properties identical to literature data.13 δH (400 MHz; CDCl3) 2.39 (3H, s), 6.09–6.49 (1H, s), 6.89 (2H, d, J = 8.0), 7.13 (2H, d, J = 8.0), 7.22 (2H, d, J = 8.0), 8.12 (2H, d, J = 8.0); δC (100.1 MHz; CDCl3) 21.0, 113.3, 122.6, 126.3, 130.2, 134.8, 136.7, 139.2 and 150.7
[N-Tolyl-p-phenylenediamine] ZnCl2 complex 5
4-Nitrophenylaminotoluene 4 (380 mg, 1.7 mmol) in EtOH (80 mL) with conc. HCl (1.5 mL) was periodically swirled on a hot plate with an excess of zinc dust (approx. 5–10 g) added in portions with a spatula (0.8 g). The conc. HCl cleans the zinc surface. The solution turned clear and was filtered hot then left to stand. Crystals of the title compound were harvested (156 mg, 31%) and characterised by an X-ray single crystal structure determination.
2,4-Diaminotoluene 6 (300 mg, 2.46 mmol) and 4-nitrofluorobenzene 2 (347 mg, 2.46 mmol) in EtOH (10 mL) were treated with Et3N (248 mg, 2.46 mmol) and heated in a pressure vessel for 24 h at 150°C. After cooling, the mixture was diluted with water (200 mL) and extracted with DCM (100 mL). The DCM layer was dried over MgSO4 and filtered. The dry extract was purified by chromatography on a silica column. Initially DCM eluted impurities then DCM/Et2O (90:10) eluted the title compound (30 mg, 5 %) an orange solid, m.p. 167–168 °C (from dichloromethane:light petroleum ether). δH (400 MHz; CDCl3) 2.28(3H, s), 6.67(1H, dd, J = 8.0 and 2.0), 6.86 (1H, d, J = 2.0), 7.15 (1H, d, J = 8.0), 7.24 (2H, d, J = 8.0) and 8.23 (2H, d, J = 8.0); δC (100.1 MHz; CDCl3) 16.6, 107.6, 110.6, 13.3, 118.4, 126.5, 131.1, 137.8, 138.7, 147.3 and 152.2; m/z (Orbitrap ASAP) 244.1088 (M + H+, 100%) C13H13N3O2 + H+ requires 244.1086.
2-Amino-4-[4-nitrophenylamino]toluene 7 (in DMSO)27,28
2,4-Diaminotoluene 6 (300 mg, 2.46 mmol) and 4-nitrofluorobenzene 2 (347 mg, 2.46 mmol) in DMSO (5 mL) were treated with Et3N (248 mg, 2.46 mmol) and heated in a round bottom flask at 90 °C for 24 h. The reaction was worked-up as above, to give the title compound (95 mg, 16%) with identical spectroscopic properties.
2-Amino-4-[4-nitrophenylamino]toluene 7 (30 mg, 0.123 mmol) and powdered N-phenyl-p-phenylenediamine 1 (23 mg, 0.123 mmol) in water (200 mL) were treated with cH2SO4 (three drops from a 15-cm Fisherbrand Pasteur pipette held at about 45°) and stirred for 1 h at 60 °C. The mixture was treated with K2Cr2O7 (36 mg, 0.123 mmol, 1 equiv.) and stirred for 2 h. Successful reactions turn a deep purple colour but this one was deep blue. After cooling, the reaction was filtered through a sinter, washed with water and extracted with MeOH (3 × 30 mL). TLC showed that no mauveine was present. Mauveine requires either 20% aqNH3:MeOH for elution or secBuOH:EtOAc:H2O:HOAc (60:30:9.5:0.5) for elution. MeOH or EtOH will not elute it.
2,4-Bis[phenylamino]aniline hydrochloride 10 was formed in situ by the zinc dust reduction of 2,4-bis[phenylamino]nitrobenzene 9 (30 mg, 0.13 mmol),31,32 filtered and eva-porated as above giving the crude hydrochloride salt. 2,4-Bis[phenylamino]nitrobenzene 931 was made by a modified method used to make compound 7 with DMSO as solvent and heated for 4 d (204 mg, 34%). 2,4-Difluoronitrobenzene (350 mg, 2.20 mmol) was heated with aniline (409 mg, 0.0044 mmol) and Et3N (445 mg, 4.40 mmol) in DMSO (5 mL) (204 mg, 34%). Data for compound 10 δH(400 MHz; CD3OD) 6.81 (1H, d, J = 8.0), 6.89 (2H, q, J = 8.0), 6.96 (2H, d, J = 8.0), 7.02 (1H, s), 7.07(2H, d, J = 8.0), 7.27-7.19(4H, m), 7.29(1H, t, J = 8.0 and 8.0); δC (100.1 MHz; CD3OD) 108.5, 111.9, 114.1, 117.3, 118.5, 120.8, 121.3, 122.8, 124.7, 129.0, 129.2, 138.6, 142.2 and 143.8. Compound 10 (26.5 mg, 0.096 mmol) and o-toluidine 11 (34 mg, 0.32 mmol) in water/acetone (200 mL:100 mL) were treated with conc. H2SO4 (three drops) was added and stirred at 60 °C in a beaker covered with a Petri dish lid. K2Cr2O7 (28 mg, 0.096 mmol) was added with stirring. After 2 h, the reaction was stirred without a lid on the beaker to evaporate acetone then cooled, filtered, washed with water and extracted in the sinter with MeOH (3 × 30 mL). TLC showed that no mauveine was present. Mauveine requires either 20% aqNH3:MeOH for elution or secBuOH:EtOAc:H2O:HOAc (60:30:9.5:0.5) for elution. MeOH or EtOH will not elute it. Salt 10 can be neutralised with aq KOH (0.1 M), extracted with DCM (50 mL) giving a light purple solution, dried over MgSO4, filtered, evaporated and chromatographed on silica. Elution with DCM then DCM/Et2O (80:20) gave 2,4-bis[phenylamino]aniline but we failed to get satisfactory NMR data. The product can be extracted because the base precipitates remaining Zn2+ ions as Zn(OH)2. If HOAc is used in place of EtOH it must be evaporated or neutralised completely as it inhibits extraction of 2,4-bis[phenylamino]aniline into DCM.
Pseudomauveine synthesis33 (3-Phenylamino-5-phenyl-7-aminophenazinium sulphate 15) Aniline trimer 1433 was prepared by the method above by in situ reduction of the nitro trimer36 using zinc dust in EtOH with small amounts of conc. HCl then filtered. The trimer (26.5 mg, 0.096 mmol) and aniline (30 mg, 0.32 mmol) in water/acetone (200 mL:100 mL) was treated with conc. H2SO4 (three drops) and stirred at 60 °C in a beaker covered with a Petri dish lid. K2Cr2O7 (28 mg, 0.096 mmol) was added with stirring. After 2 h, the reaction was cooled, filtered, washed with water and extracted in the sinter with MeOH (3 × 30 mL). The MeOH extract was evaporated and purified by chromatography. Elution with MeOH then aq NH3:MeOH (20:80) gave the title compound (17 mg, 45%) with identical spectroscopic properties to that reported previously. δH(400 MHz; CDCl3) 5.91 (1H, d, J = 1.6), 6.22 (1H, d, J = 1.6), 7.02 (1H, m), 7.05 (2H, d, J = 7.4), 7.20 (3H, m), 7.35 (1H, m), 7.41 (2H, d, J = 7.4), 7.65 (1H, d, J = 7.4), 7.71 (2H, m), 7.79 (1H, d, J = 9) and 7.86 (1H, d, J = 9.4); δC(100.1 MHz; CDCl3) 93.2, 93.7, 120.3, 121.7, 121.8, 125.0, 127.2, 129.0, 130.6, 131.0, 133.0, 133.7, 135.9, 136.1, 136.4, 137.2, 137.5, 138.4, 152.5 and 158.1
Synthesis of phenyliminoquinone 1723 (failed trimer synthesis 1)
N-Phenyl-p-phenylenediamine 1 (300 mg, 1.63 mmol) and 2,6-dimethylaniline 16 (197 mg, 1.63 mmol) in water (300 mL) was treated with cH2SO4 (6 drops from a 15- cm Pasteur pipette) then K2Cr2O7 (320 mg, 0.66 equiv.) and heated at 60 °C with stirring for 2 h. After cooling the mixture was filtered and washed with water. The product in the sinter was extracted with MeOH (3 × 50 mL) and evaporated. The product was purified by chromatography on silica. Elution with MeOH gave the title compound (139 mg, 47%) of identical spectroscopic properties to literature material. δH (400 MHz; CD3OD) 6.92–6.96 (3H, m), 7.15–7.18 (4H, m) and 7.29 (2H, t, J = 9.0 and 7.0); δC (100.1 MHz; CD3OD) 116.0, 118.2, 120.8, 124.3, 128.9, 141.2, 142.4, 144.1, 155.8 and 188.2. m/z (Orbitrap ASAP) 184.0763 (M + H+, 100%) C12H9NO + H+ requires 184.0762.
Synthesis of phenyliminoquinone (failed trimer synthesis 2)
The above reaction was repeated using N,N-dimethylaniline 18, which is an isomer of 2,6-dimethylaniline 16. The product had identical properties to that reported above (156 mg, 52%).
N-Phenyl-p-phenylenediamine 1 (300 mg, 1.63 mmol) in water (300 mL) was treated with cH2SO4 (four drops from a 15-cm Pasteur pipette) and then K2Cr2O7 (160 mg, 1/3 equiv.) was added and heated at 60 °C with stirring for 2 h. After cooling, the mixture was filtered and washed with water. The product in the sinter was extracted with MeOH (3 × 50 mL) and evaporated. TLC showed that no blue product was present. The plate was photographed (Figure 13).
Crystal structure determinations
The crystal structures of compound 5 (colourless block 0.20 × 0.10 × 0.10 mm) and 7 (orange block 0.30 × 0.15 × 0.10 mm) were established using intensity data collected on a Rigaku CCD diffractometer. The structures were routinely solved by dual-space methods using SHELXT37 and the structural models were completed and optimised by refinement against |F|2 with SHELXL.38 The N-bound H atoms were located in difference maps and their positions were freely refined. The C-bound H atoms were placed in idealised locations (C–H = 0.95–0.98 Å) and refined as riding atoms. The methyl groups were allowed to rotate, but not to tip, to best fit the electron density. The constraint Uiso(H) = 1.2Ueq(carrier) or 1.5Ueq(methyl carrier) was applied in all cases. Full details of the structures and refinements are available in the deposited cifs.
Crystal data for compound 5 C26H28Cl2N4Zn, Mr = 532.79, monoclinic, space group Ia (No. 9), a = 8.9096 (4) Å, b = 5.9680 (3) Å, c = 44.852 (3) Å, β = 92.401 (5)°, V = 2382.8 (3) Å3, Z = 4, T = 100 K, Mo Kα radiation, λ = 0.71073 Å, μ = 1.278 mm–1, ρcalc = 1.485 g cm–3, 7163 reflections measured (3.7 ⩽ 2θ ⩽ 55.0°), 4150 unique (RInt = 0.053), R(F) = 0.059 [3380 reflections with I > 2σ(I)], wR(F2) = 0.135 (all data), Flack absolute structure parameter = 0.00 (2), Δρmin,max (e Å–3) = –0.56, +0.94, CCDC deposition number 2425924.
Crystal data for compound 7 C13H13N3O2, Mr = 243.26, orthorhombic, space group Pbca (No. 61), a = 14.5556 (5) Å, b = 6.5516 (2) Å, c = 24.2152 (9) Å, V = 2309.22 (14) Å3, Z = 8, T = 100 K, Mo Kα radiation, λ = 0.71073 Å, μ = 0.097 mm–1, ρcalc = 1.399 g cm–3, 35,542 reflections measured (3.4 ⩽ 2θ ⩽ 61.0°), 3522 unique (RInt = 0.029), R(F) = 0.038 [3061 reflections with I > 2σ(I)], wR(F2) = 0.108 (all data), Δρmin,max (e Å–3) = –0.21, +0.41, CCDC deposition number 2425925.
Supplemental Material
sj-docx-1-chl-10.1177_17475198251347334 – Supplemental material for Failed mauveine syntheses and the mechanistic insight they provide into the role of linear trimers in mauveine synthesis
Supplemental material, sj-docx-1-chl-10.1177_17475198251347334 for Failed mauveine syntheses and the mechanistic insight they provide into the role of linear trimers in mauveine synthesis by Michael J Plater and William TA Harrison in Journal of Chemical Research
Footnotes
Acknowledgements
We thank the UK EPSRC National Mass Spectrometry Service Centre for mass spectrometric data and the UK National Crystallography Service Centre (University of Southampton) for the X-ray data collections. M John Plater performed all synthesis and obtained the characterisation data and WTA Harrison solved the crystallographic data sets. Data sets were obtained free of charge from the National Crystallography Service Centre, University of Southampton.
ORCID iD
Michael J Plater
Ethical considerations
Ethical approval was not required for this project. WTA Harrison has given approval for this publication.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Data availability statement
This paper and the supplementary will be made available in the Aberdeen University Duncan Rice library,
Supplemental material
Supplemental material for this article is available online.
References
1.
PerkinWH.PatentGB1856, 26August, No 1984.
2.
DaleJCaroH.PatentGB1860, 26May, No 1307.
3.
PerkinWH.J Chem Soc1879; 35: 717–732.
4.
MansfieldCB.PatentGB1847, No 11,960.
5.
GardnerWM.The British coal tar industry: Its origin, development and decline. Philadelphia, PA: J B Lippincott Company, 1915.
6.
FindlayA.The treasures of coal tar. London: Forgotten Books, 2018.
7.
ProQuest. From coal to colour: once a week. CooperT (ed.) Mark Lemon. London: ProQuest, 1871, 194–197.
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.