
Abstract
In order to mitigate calcium carbonate (CaCO3) scaling in oil and gas wells, antiscaling agents are commonly used. This research employed the molecular dynamics method to simulate the crystallization process of a supersaturated CaCO3 solution, with particular emphasis on examining the influence of the antiscalant edetate sodium (EDTA). Analysis of the spatial distribution of Ca2+ in proximity to CO32− revealed that EDTA effectively impedes the interaction between Ca2+ and CO32−, resulting in a reduction in the mean square displacement and diffusion coefficient of CO32−. The examination of intermolecular interaction energies indicates that the binding energy between Ca2+ and CO32− is approximately −516 kcal mol−1, while the binding energy between Ca2+ and EDTA is estimated at −718 kcal mol−1. These findings suggest that EDTA exhibits a higher affinity for binding with Ca2+ compared to CO32−, consequently hindering the formation of CaCO3.
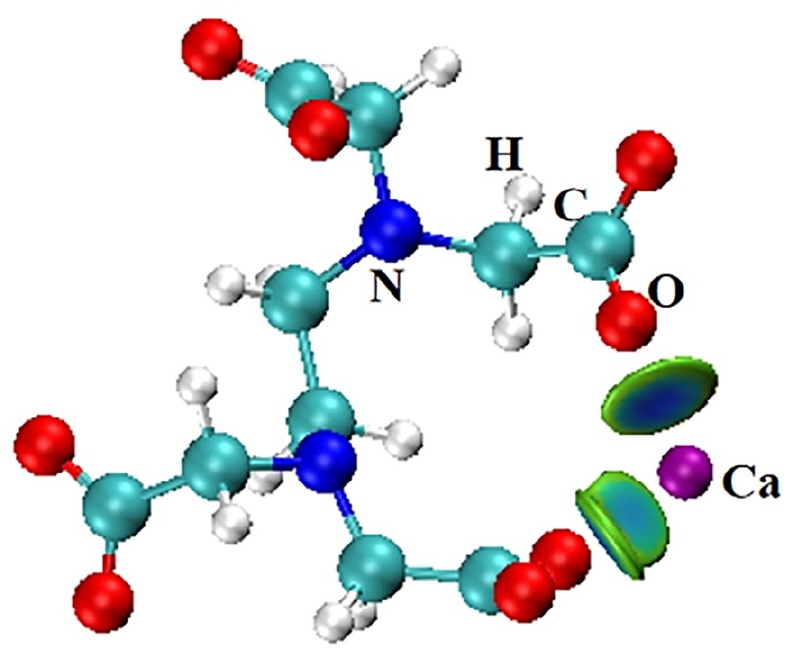
Schematic diagram of EDTA molecular complexation with calcium ions
Introduction
In Moxi Gas Field in Sichuan, the Longwangmiao formation gas wells exhibit a high salinity water production capacity, and multiple scaling ions can be detected in the formation water. 1 The occurrence of scale blockage in oil pipes, primarily in the form of calcium carbonate (CaCO3) scale, frequently leads to a notable reduction in gas well productivity,2,3 and the application of hydrochloric acid is the most common strategy for removing carbonate deposits.4,5 However, implementation of this strategy results in increased levels of carbon dioxide (CO2) and hydrogen sulfide (H2S) in the gas, along with significant corrosion to metal pipes. 6 In addition, frequent blockage of wellbores results in costly removal procedures, as well as production interruptions. To control blockages caused by the CaCO3 scaling, the addition of scale inhibitors to aqueous solutions is often used.7,8
Phosphate is a kind of high-performance scale inhibitor. 9 However, due to its potential environmental harm, research is increasingly concentrated on the development of environmentally friendly scale inhibitors. In the work by Chen et al., 10 a novel and highly efficient polyaspartic acid/graphene oxide grafted copolymer (PASP/GO) was prepared. They evaluated the scale inhibition performance of PASP/GO and studied the mechanism by using the molecular dynamics (MD) simulation method. According to the results, PASP/GO exhibited excellent inhibition performance and dispersion capacity, and PASP/GO adsorbs scale particles by chelation, crystal distortion, and dispersion. In the work by Zuo et al., 11 they studied the performance of several common scale inhibitors by using MD methods and demonstrated that 2-phosphanobutane-1,2,4-tricarboxylic acid (PBTCA) has the best scale inhibition performance.
In the work by Zeng et al., 12 MD simulation was conducted to investigate the interaction between various configurations of poly(aspartic acid) (PASP) and different surfaces of CaCO3 crystals in aqueous solution. The findings indicate that PASP effectively inhibits the growth of CaCO3 scale, with the binding energies primarily arising from coulombic interactions, including ionic bonds. In the work by Zhang et al., 13 experimental and MD simulation methodologies were both used to meticulously investigate the scale inhibitory properties of carboxymethyl dextran (CMD) on the nucleation and crystallization kinetics of CaCO3. The findings revealed significant interactions between Ca2+ in solution and the carboxyl groups of CMD. As the concentration of CMD increased, the likelihood and strength of adsorption of Ca2+ and CO32− onto the solution’s surface decreased.
Due to environmental concerns, phosphorus-based scale inhibitors are not included in our study. Edetate sodium (EDTA) serves as a scale inhibitor whose functional groups can adsorb Ca2+ in industrial circulating water.2,14,15 To investigate the fundamental principles underlying the function of EDTA as a scale inhibitor, this study examines the ion aggregation in CaCO3 solution through MD simulation with and without EDTA; additionally, weak interactions between ions were also evaluated. The findings suggest that the interaction energy between EDTA and Ca2+ surpasses that of CO32− and Ca2+, indicating a higher likelihood of EDTA binding with Ca2+ and preventing the precipitation of CaCO3.
Computational details
In this study, GROMACS software version 2022.516,17 was employed to simulate the aggregation of Ca2+ and CO32− ions with and without the scale inhibitor EDTA, the molecular structure formula is shown in Figure 1. The General AMBER Force Field (GAFF) was used for the characterization of small molecules, 18 with atomic charges being assigned as the advanced restrained electrostatic potential (RESP2) charges. 19 The ion parameters were obtained from existing literature sources,20,21 and OPC3 water model 22 was used to describe water. Packmol software 23 was used to build the simulation box with initial size of 10 nm × 10 nm × 10 nm. Within the control group, the concentration of the scale inhibitor EDTA is approximately 3%, the number of molecules and atoms present in the two systems are listed in Table 1. During the simulation, the temperature was set at 320 K and the pressure at 10 MPa in order to mimic the conditions in the wellbore. Following the minimization of system energy utilizing the steepest descent algorithm, leapfrog algorithm was used to perform 100 ns MD simulation under the canonical ensemble with constant pressure and temperature (NPT) ensemble; Parrinello-Rahman algorithm 24 with pressure coupling constant time of 2.5 ps and the V-rescale thermostat algorithm 25 with coupling constant time of 2 ps were both used to control the pressure and temperature within the simulation system. The Coulomb interaction calculation method was particle-mesh Ewald (PME) algorithm 26 and the van der Waals interaction algorithm was cut-off; the cutoff were both 1 nm. Visual molecular dynamics (VMD 1.9.3) software 27 was used for visual observation.

Molecular structural formula of EDTA (structural and ball-stick model).
The number of molecules and atoms in the simulation box.

Independent gradient model based on Hirshfeld partition (IGMH) analysis 28 steps: first, select all atoms/molecules within 3.5 nm radius of a specified atom/molecule in VMD, followed by the utilization of ORCA 5.0.429–31 for the acquisition of wavefunction data at the B3LPY D3 ma-def2-TZVP level. Subsequently, Multiwfn 32 was used for IGMH analysis based on the obtained wavefunction information.
Results and discussion
Figure 2(a) and (c) depict the initial state prior to MD simulation with and without the scale inhibitor EDTA, respectively, showing all molecules/atoms in a disordered arrangement. In contrast, Figure 2(b) and (d) illustrate the equilibrium state, where molecules/atoms aggregated together due to electrostatic attraction between anions and cations. The trajectory videos can be accessed at the following URLs https://youtu.be/IUDTrv2TSDk and https://youtu.be/dsZkiOZBqAM. There is a notable disparity in the distribution of molecules/atoms before and after simulation, with the aggregation of molecules/atoms after equilibrium showing an area of interest for further investigation.

Screenshots before and after MD simulation (: Ca2+; : CO32+;
CO32+;  : EDTA;
: EDTA;  : Na+).
: Na+).
The radial distribution function (RDF) is capable of characterizing the spatial arrangement of Ca2+ and CO32+. It indicates the position of a given particle

where
Figure 3 shows the RDF analysis results. The peak occurrence of Ca2+ ions at a distance of 0.23 nm from CO32− ions suggests the formation of the first coordination layer. A minor peak is observed at approximately 0.38 nm, which is believed to be associated with the second coordination layer. From the perspective of peak intensity, the second coordination layer can be ignored. This observation suggests that CO32− and Ca2+ ions exhibit a tendency to form pairs due to the opposing charges attraction, but not all anions are capable of pairing with cations. In contrast, the presence of EDTA in the solution leads to a significant decrease in the RDF value between Ca2+ and CO32−, suggesting that the interaction between Ca2+ and CO32− is impeded, thereby inhibiting the binding of Ca2+ and CO32−. This phenomenon also elucidates the mechanism by which scale inhibitors function.
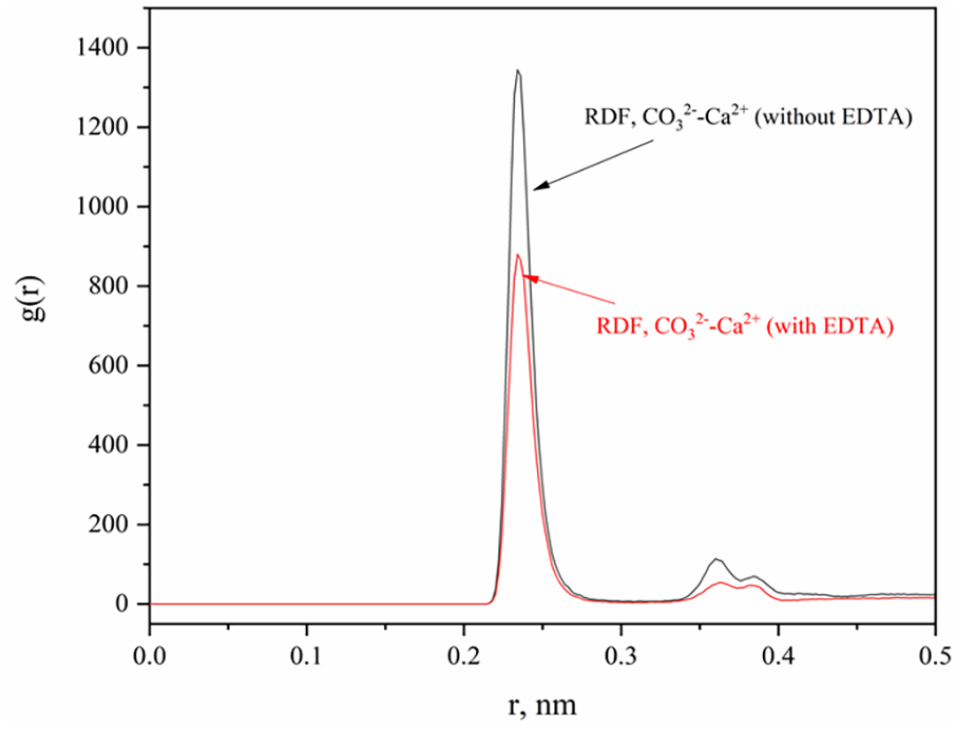
RDF of Ca2+ and CO32− with and without EDTA.
Based on Figure 3, it is evident that Ca2+ and CO32− exhibit the greatest likelihood of being situated at a distance of 0.23 nm. To determine the precise quantity in the MD process, we quantified the number of CO32− ions in close proximity to Ca2+ ions, utilizing a distance threshold of 0.25 nm, marginally exceeding the 0.23 nm distance observed in Figure 3.
According to Figure 4, in the system without scale inhibitor, Ca2+ ions and CO32− ions exhibit rapid aggregation as soon as the simulation begins, with an average of 6.5 CO32− ions in close proximity to each Ca2+ ion when reaching equilibrium. This observation aligns with the characteristic structure of CaCO3 crystals. 33 Conversely, in the system with scale inhibitor EDTA, the proximity of CO32− ions to Ca2+ ions is significantly reduced, with only about 3 CO32− ions in close proximity to each Ca2+ ion when reaching equilibrium, suggesting that the inhibitor effectively hinders the formation of CaCO3 crystals.

Number of Ca2+ near 0.25 nm of CO32−.
In the course of MD simulations, particles within the system exhibit continuous movement from their initial positions, and their positions are different at each moment. The quantification of this displacement is commonly referred to as the mean square displacement (MSD). The MSD is able to assess the extent to which the position of a particle diverges from its initial state over a defined duration. In this study, we used MSD to quantify the movement of Ca2+ and CO32−. The degree of deviation of the positions of these molecules from their initial reference positions is determined over time through the application of equation (1), where

Figure 5 demonstrates that the MSD curves of Ca2+ and CO32− ions without scale inhibitor exhibit close proximity, suggesting a consistent pairing of these ions; this is caused by the attraction of charges, and the diffusion coefficient is very close, about 0.3769 cm2 s−1. On the other hand, the presence of scale inhibitors in the solution results in a notable decrease in the diffusion coefficient of Ca2+ and CO32− ions, and it is about 0.1745 cm2 s−1. This reduction in diffusion coefficient indicates that EDTA impedes the mobility of these ions, thereby hindering the formation and growth of CaCO3 crystals.
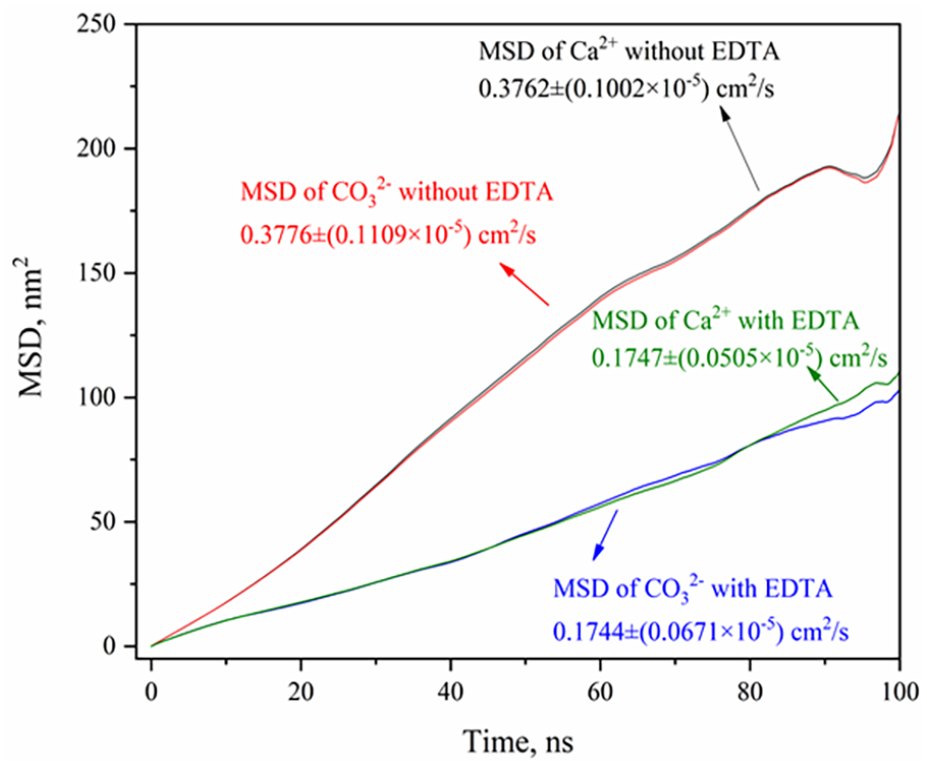
MSD of Ca2+ and CO32− in the two systems.
The weak interactions between molecules/atoms are also a very worthwhile aspect to study. In 2022, Lu introduced a simple and intuitive method, known as IGMH, 28 to study the weak interactions between molecules/atoms. Following the simulation of equilibrium, our research primarily focuses on exploring the interactions between ions. Of particular interest are the interactions between Ca2+ and CO32− ions, as well as those between EDTA and Ca2+ ions.
As presented in Figure 6, there are two kinds of weak interactions in the IGMH analysis graphs, appearing as blue and green isosurfaces. There are several very clear blue isosurface between Ca2+ and CO32− ions; unquestionably, they are attributed to the attraction of opposite charges. The green isosurfaces between CO32− indicate the existence of van der Waals interactions. It should be noted that a crystal-like configuration is formed between Ca2+ and CO32−; please refer to the following URL for observation: https://youtu.be/IgpQ9f_bAIM. On the other hand, the isosurface between EDTA and Ca2+ ions is also blue, indicating that it is also formed by opposite charge attraction. It should be noted that both carboxylate groups in EDTA bind to the same Ca2+.

IGMH analysis of weak intermolecular interactions.
From the colors of the isosurface in Figure 6, it seems that the attraction between CO32− and Ca2+ is stronger than that between EDTA and Ca2+. This contradicts the theory of EDTA as a scale inhibitor. Quantitative assessment of interaction strength serves as a key indicator for evaluating binding stability between molecules or ions. The method of energy decomposition is used to provide a quantitative characterization of the various sources of intermolecular interactions. To investigate the interaction energy between ions, the PSI4 code 34 was used for high-order symmetry-adapted perturbation theory (SAPT) analyses. Because Ca belongs to the fourth period element, and due to our limited computing resources, we cannot use extremely advanced base sets aug-cc-pVTZ, the def2-TZVPP basis set was used, with the keywords SAPT2+(3) δ MP2/def2-TZVPP. The SAPT input files of PSI4 were generated with the help of Multiwfn program. 32
From the data presented in Table 2, it is evident that the binding energy between ions is negative, suggesting that the attraction between these species occurs spontaneously without the need for an external driving force. Further analysis through energy decomposition reveals that electrostatic interactions predominantly contribute to this phenomenon, underscoring the spontaneous nature of the mutual attraction between anions and cations. It can be inferred that the interaction energy between EDTA and Ca2+ ions is greater than that between CO32− and Ca2+ ions, indicating a higher propensity for Ca2+ ions to bind with EDTA and thereby prevent the precipitation of CaCO3. This fundamental principle of utilizing EDTA as a scale inhibitor is also applicable in the research and development of novel scale inhibitors.
Analysis of interaction energy between ions (kcal mol−1).

Conclusion
Based on MD simulations, this study investigates how EDTA works as a scale inhibitor. Through the application of the IGMH method, this study illustrated the weak interactions between CO32− ions, EDTA, and Ca2+ ions, revealing that mutual attraction is predominantly driven by charge interactions. In the presence of scale inhibitors EDTA, a significant reduction in the number of CO32− ions in close proximity to Ca2+ ions was observed. The energy decomposition method was used to quantify ion interaction strength. The findings indicated that the interaction energy between EDTA and Ca2+ ions approached −718 kcal mol−1, whereas the interaction energy between CO32− and Ca2+ ions was approximately −516 kcal mol−1. Evidently, EDTA exhibits a greater affinity for Ca2+ ions, impeding the association between Ca2+ ions and CO32− ions, thereby inhibiting the formation of CaCO3 crystals.
Footnotes
Authors’ Note
X.J. and B.Z. wrote the main manuscript text, and L.Z. and F.F. prepared Figures 1–![]() and Y.L. polished the language. All authors reviewed the manuscript.
and Y.L. polished the language. All authors reviewed the manuscript.
Availability of data and materials
All the data that support the findings of this study are available on request from the corresponding author.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Sichuan Provincial Science and Technology Plan Project (2023YFG0092).
Consent for publication
The authors declare that they have no known competing financial interests or personal relationships that may affect the work reported in this paper.